Sarkoidose
| Sarkoidose ⓘ | |
|---|---|
| Andere Namen | Sarkoidose, Sarkoid, Besnier-Boeck-Schaumann-Krankheit |
 | |
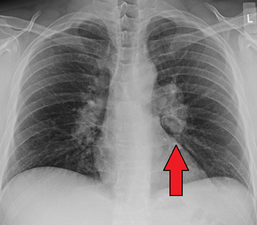
| Röntgenbild des Brustkorbs, das die für die Sarkoidose typische Knötchenbildung zeigt, vor allem in den Lungenbasen. | |
| Aussprache |
|
| Fachgebiet | Rheumatologie, Immunologie |
| Symptome |
|
| Gewöhnlicher Ausbruch | 20-50 Jahre alte Frauen |
| Dauer | Wenige Jahre bis lang anhaltend |
| Ursachen | Unbekannt |
| Risikofaktoren | Familienanamnese |
| Diagnostische Methode | Anhand der Symptome und einer Gewebebiopsie |
| Differentialdiagnose | Tuberkulose, Lymphom, infektiöse Mononukleose, pulmonale Eosinophilie |
| Behandlung | Ibuprofen, Prednison, Methotrexat |
| Prognose | Sterblichkeit 1-7% |
| Häufigkeit | 1,9 Millionen Menschen mit interstitieller Lungenerkrankung (2015) |
| Todesfälle | 122.000 mit interstitieller Lungenerkrankung (2015) |
Sarkoidose (auch als Besnier-Boeck-Schaumann-Krankheit bekannt) ist eine Krankheit, bei der sich abnorme Ansammlungen von Entzündungszellen bilden, die als Granulome bezeichnet werden. Die Krankheit beginnt meist in der Lunge, der Haut oder den Lymphknoten. Seltener sind die Augen, die Leber, das Herz und das Gehirn betroffen. Es kann aber auch jedes andere Organ betroffen sein. Die Anzeichen und Symptome hängen von dem betroffenen Organ ab. Oft treten keine oder nur leichte Symptome auf. Wenn die Lunge betroffen ist, können Keuchen, Husten, Kurzatmigkeit oder Brustschmerzen auftreten. Bei einigen Patienten kann das Löfgren-Syndrom mit Fieber, großen Lymphknoten, Arthritis und einem als Erythema nodosum bezeichneten Ausschlag auftreten. ⓘ
Die Ursache der Sarkoidose ist unbekannt. Manche glauben, dass sie bei genetisch Veranlagten auf eine Immunreaktion auf einen Auslöser wie eine Infektion oder Chemikalien zurückzuführen ist. Personen mit betroffenen Familienmitgliedern sind einem höheren Risiko ausgesetzt. Die Diagnose basiert zum Teil auf Anzeichen und Symptomen, die durch eine Biopsie bestätigt werden können. Zu den Befunden, die die Diagnose wahrscheinlich machen, gehören große Lymphknoten an der Lungenwurzel auf beiden Seiten, ein hoher Kalziumspiegel im Blut bei normalem Nebenschilddrüsenhormonspiegel oder ein erhöhter Spiegel des Angiotensin-konvertierenden Enzyms im Blut. Die Diagnose sollte erst nach Ausschluss anderer möglicher Ursachen für ähnliche Symptome, wie z. B. Tuberkulose, gestellt werden. ⓘ
Die Sarkoidose kann sich ohne Behandlung innerhalb weniger Jahre zurückbilden. Bei manchen Menschen kann die Krankheit jedoch langwierig oder schwer verlaufen. Einige Symptome können durch die Einnahme von entzündungshemmenden Medikamenten wie Ibuprofen gelindert werden. In Fällen, in denen die Erkrankung erhebliche gesundheitliche Probleme verursacht, sind Steroide wie Prednison angezeigt. Gelegentlich können Medikamente wie Methotrexat, Chloroquin oder Azathioprin eingesetzt werden, um die Nebenwirkungen der Steroide zu verringern. Das Sterberisiko liegt bei 1-7 %. Die Wahrscheinlichkeit, dass die Krankheit bei jemandem, der sie schon einmal hatte, zurückkehrt, liegt bei weniger als 5 %. ⓘ
Im Jahr 2015 waren weltweit 1,9 Millionen Menschen an pulmonaler Sarkoidose und interstitieller Lungenerkrankung erkrankt, und es gab 122 000 Todesfälle. Sie ist in Skandinavien am häufigsten, kommt aber in allen Teilen der Welt vor. In den Vereinigten Staaten ist das Risiko bei Schwarzen höher als bei Weißen. Die Krankheit beginnt in der Regel im Alter zwischen 20 und 50 Jahren. Sie tritt häufiger bei Frauen als bei Männern auf. Die Sarkoidose wurde erstmals 1877 von dem englischen Arzt Jonathan Hutchinson als eine nicht schmerzhafte Hautkrankheit beschrieben. ⓘ
| Klassifikation nach ICD-10 ⓘ | |
|---|---|
| D86.0 | Sarkoidose der Lunge |
| D86.1 | Sarkoidose der Lymphknoten |
| D86.2 | Sarkoidose der Lunge mit Sarkoidose der Lymphknoten |
| D86.3 | Sarkoidose der Haut |
| D86.8 | Sarkoidose an sonstigen und kombinierten Lokalisationen |
| D86.9 | Sarkoidose, nicht näher bezeichnet |
| ICD-10 online (WHO-Version 2019) | |


Die Sarkoidose (von altgriechisch σαρκωειδής sarkoeidés „fleischartig, fleischig“), auch als Morbus Boeck (buːk), Boecksche Krankheit oder Morbus Schaumann-Besnier bezeichnet, ist eine systemische Erkrankung des Bindegewebes mit Granulombildung, die meistens zwischen dem 20. und 40. Lebensjahr auftritt. Die genaue Ursache der Krankheit ist bis heute unbekannt. ⓘ
Bei der Sarkoidose bilden sich mikroskopisch kleine Knötchen (Granulome) in dem betroffenen Organgewebe, verbunden mit einer verstärkten Immunantwort. ⓘ
Man unterscheidet eine zunächst akut verlaufende Form der Sarkoidose, das sogenannte Löfgren-Syndrom, von der schleichend und symptomarm einsetzenden chronischen Verlaufsform. In Deutschland tritt die Sarkoidose in 20 bis 30 Fällen auf 100.000 Einwohner auf. ⓘ
Erstmals war sie von Ernest Besnier und Cæsar Peter Møller Boeck in den Jahren 1889 und 1899 als Hauterkrankung beschrieben worden. Im Jahre 1924 erkannte Jörgen Nilsen Schaumann, dass es sich hierbei um eine systemische Erkrankung verschiedener Organe handelt. Der Schwede Sven Halvar Löfgren beschrieb 1953 die nach ihm benannte akute Verlaufsform. ⓘ
Anzeichen und Symptome


Die Sarkoidose ist eine systemische Entzündungskrankheit, die alle Organe befallen kann. Sie kann jedoch auch asymptomatisch sein und wird in etwa 5 % der Fälle zufällig entdeckt. Zu den häufigen, eher vagen Symptomen gehören Müdigkeit (die sich auch durch Schlaf nicht bessert; tritt in bis zu 85 % der Fälle auf), Energielosigkeit, Gewichtsverlust, Gelenkschmerzen (die in etwa 70 % der Fälle auftreten), Arthritis (14-38 % der Fälle), trockene Augen, geschwollene Knie, verschwommenes Sehen, Kurzatmigkeit, trockener, hackender Husten oder Hautveränderungen. Seltener können die Betroffenen Blut aushusten. Sarkoidose geht auch mit psychischen Problemen und Symptomen von Angst und Depression einher, die auch mit Müdigkeit verbunden sind. Die Hautsymptome sind unterschiedlich und reichen von Hautausschlägen und Noduli (kleinen Beulen) bis hin zu Erythema nodosum, Granuloma annulare oder Lupus pernio. Sarkoidose und Krebs können sich gegenseitig imitieren, was die Unterscheidung erschwert. ⓘ
Die Kombination aus Erythema nodosum, bilateraler hilärer Lymphadenopathie und Gelenkschmerzen wird als Löfgren-Syndrom bezeichnet, das eine relativ gute Prognose hat. Diese Form der Erkrankung tritt bei skandinavischen Patienten deutlich häufiger auf als bei Patienten nicht-skandinavischer Herkunft. ⓘ
Atmungsorgane
Die Lokalisierung in der Lunge ist die bei weitem häufigste Erscheinungsform der Sarkoidose. Bei mindestens 90 % der Betroffenen ist die Lunge betroffen. Insgesamt entwickeln etwa 50 % dauerhafte pulmonale Anomalien, und 5 bis 15 % haben eine fortschreitende Fibrose des Lungenparenchyms. Die Sarkoidose der Lunge ist in erster Linie eine interstitielle Lungenerkrankung, bei der der Entzündungsprozess die Alveolen, die kleinen Bronchien und die kleinen Blutgefäße betrifft. In akuten und subakuten Fällen zeigt die körperliche Untersuchung in der Regel trockene Knistergeräusche. Mindestens 5 % der Fälle weisen eine pulmonal-arterielle Hypertonie auf. Die oberen Atemwege (einschließlich Kehlkopf, Rachen und Nasennebenhöhlen) können betroffen sein, was in 5 bis 10 % der Fälle der Fall ist. ⓘ
Die vier Stadien der Lungenbeteiligung richten sich nach dem radiologischen Stadium der Erkrankung, das für die Prognose hilfreich ist:
- Stadium I: bilaterale hiläre Lymphadenopathie (BHL) allein
- Stadium II: BHL mit pulmonalen Infiltraten
- Stadium III: pulmonale Infiltrate ohne BHL
- Stadium IV: Fibrose ⓘ
Die Verwendung der Scadding-Skala liefert nur allgemeine Informationen über die Prognose der Lungenerkrankung im Zeitverlauf. Es wird zur Vorsicht geraten, da die Skala nur eine allgemeine Beziehung zu physiologischen Markern der Krankheit aufzeigt und die Schwankungen so groß sind, dass sie für individuelle Beurteilungen, einschließlich Behandlungsentscheidungen, nur begrenzt geeignet ist. ⓘ

Als Faustregel gilt: Je jünger der Patient, je akuter der Verlauf, desto besser die Prognose. ⓘ
Haut
Die Haut ist in 9 bis 37 % der Fälle von Sarkoidose betroffen und kommt bei Afroamerikanern häufiger vor als bei europäischen Amerikanern. Die Haut ist nach der Lunge das am zweithäufigsten betroffene Organ. Die häufigsten Läsionen sind Erythema nodosum, Plaques, makulopapulöse Eruptionen, subkutane Knötchen und Lupus pernio. Eine Behandlung ist nicht erforderlich, da sich die Läsionen in der Regel innerhalb von 2-4 Wochen spontan zurückbilden. Obwohl sie entstellend sein kann, verursacht die kutane Sarkoidose selten größere Probleme. Sarkoidose der Kopfhaut äußert sich in diffusem oder fleckigem Haarausfall. ⓘ
Herz
Histologisch gesehen handelt es sich bei der Sarkoidose des Herzens um eine aktive granulomatöse Entzündung, die von einem reaktiven Ödem umgeben ist. Die Verteilung der betroffenen Bereiche ist lückenhaft und die Herzmuskeln sind lokalisiert vergrößert. Dies führt zu einer Vernarbung und einem Umbau des Herzens, was zu einer Erweiterung der Herzhöhlen und einer Ausdünnung der Herzmuskulatur führt. Im weiteren Verlauf kommt es zu einem Aneurysma der Herzkammern. Bei einer diffusen Verteilung kommt es zu einer Dilatation beider Herzkammern, was zu Herzversagen und Herzrhythmusstörungen führt. Wenn das Reizleitungssystem in der Herzscheidewand betroffen ist, führt dies zu Herzblockaden, ventrikulären Tachykardien und ventrikulären Arrhythmien, die zum plötzlichen Tod führen. Die Beteiligung des Herzbeutels und der Herzklappen ist jedoch ungewöhnlich. ⓘ
Die Häufigkeit der Herzbeteiligung variiert und wird erheblich von der Rasse beeinflusst; in Japan haben mehr als 25 % der Sarkoidosepatienten eine symptomatische Herzbeteiligung, während in den USA und Europa nur etwa 5 % der Fälle eine Herzbeteiligung aufweisen. Autopsiestudien in den USA haben eine Häufigkeit der Herzbeteiligung von etwa 20-30 % ergeben, während Autopsiestudien in Japan eine Häufigkeit von 60 % ergaben. Das Erscheinungsbild der kardialen Sarkoidose kann von asymptomatischen Überleitungsanomalien bis hin zu tödlichen ventrikulären Arrhythmien reichen. ⓘ
Überleitungsanomalien sind die häufigsten kardialen Manifestationen der Sarkoidose beim Menschen und können bis zum kompletten Herzblock reichen. An zweiter Stelle der Häufigkeit von Überleitungsanomalien stehen ventrikuläre Arrhythmien, die in etwa 23 % der Fälle mit Herzbeteiligung auftreten. Der plötzliche Herztod, entweder aufgrund ventrikulärer Arrhythmien oder eines vollständigen Herzblocks, ist eine seltene Komplikation der kardialen Sarkoidose. Die kardiale Sarkoidose kann eine Fibrose, eine Granulombildung oder eine Flüssigkeitsansammlung im Interstitium des Herzens oder eine Kombination der beiden ersteren verursachen. Die kardiale Sarkoidose kann auch zu kongestiver Herzinsuffizienz führen, wenn Granulome eine Myokardfibrose und Narbenbildung verursachen. Eine kongestive Herzinsuffizienz tritt bei 25-75 % der Personen mit kardialer Sarkoidose auf. Diabetes mellitus und sarkoidosebedingte Herzrhythmusstörungen gelten als starke Risikofaktoren für Herzversagen bei Sarkoidose. Ein geringes (20-40 %) erhöhtes Risiko für einen akuten Myokardinfarkt wurde ebenfalls beschrieben. Die pulmonal-arterielle Hypertonie tritt bei der kardialen Sarkoidose durch zwei Mechanismen auf: eine eingeschränkte Linksherzfunktion aufgrund von Granulomen, die den Herzmuskel schwächen, oder aufgrund eines beeinträchtigten Blutflusses. ⓘ
Auge
Die Augen sind in etwa 10-90 % der Fälle betroffen. Zu den Manifestationen am Auge gehören Uveitis, Uveoparotitis und Netzhautentzündungen, die zum Verlust der Sehschärfe oder zur Erblindung führen können. Die häufigste ophthalmologische Manifestation der Sarkoidose ist die Uveitis. Die Kombination aus anteriorer Uveitis, Parotitis, Lähmung des VII. Hirnnervs und Fieber wird als uveoparotides Fieber oder Heerfordt-Syndrom bezeichnet (D86.8). Die Entwicklung von Skleraknötchen im Zusammenhang mit einer Sarkoidose ist beobachtet worden. ⓘ
Nervensystem
Alle Komponenten des Nervensystems können betroffen sein. Sarkoidose, die das Nervensystem betrifft, wird als Neurosarkoidose bezeichnet. Am häufigsten sind Hirnnerven betroffen, die etwa 5-30 % der Neurosarkoidosefälle ausmachen, und eine periphere Gesichtsnervenlähmung, oft beidseitig, ist die häufigste neurologische Manifestation der Sarkoidose. Sie tritt plötzlich auf und ist in der Regel vorübergehend. Das zentrale Nervensystem ist in 10-25 % der Sarkoidosefälle betroffen. Weitere häufige Manifestationen der Neurosarkoidose sind Funktionsstörungen des Sehnervs, Papillenödeme, Gaumenfunktionsstörungen, neuroendokrine Veränderungen, Hörstörungen, hypothalamische und hypophysäre Anomalien, chronische Meningitis und periphere Neuropathie. Eine Myelopathie, d. h. eine Beteiligung des Rückenmarks, tritt in etwa 16-43 % der Neurosarkoidosefälle auf und ist häufig mit der schlechtesten Prognose der Neurosarkoidose-Subtypen verbunden. Während Gesichtsnervenlähmungen und akute sarkoidosebedingte Meningitis in der Regel die günstigste Prognose haben, ist ein weiterer häufiger Befund bei Sarkoidose mit neurologischer Beteiligung eine autonome oder sensorische Kleinfaserneuropathie. Die neuroendokrine Sarkoidose macht etwa 5-10 % der Neurosarkoidosefälle aus und kann zu Diabetes insipidus, Veränderungen des Menstruationszyklus und hypothalamischen Funktionsstörungen führen. Letztere kann zu Veränderungen der Körpertemperatur, der Stimmung und des Prolaktinspiegels führen (Einzelheiten siehe Abschnitt Endokrinologie und Exokrinologie). ⓘ
Endokrin und exokrin
Prolaktin ist bei Sarkoidose häufig erhöht, zwischen 3 und 32 % der Fälle haben eine Hyperprolaktinämie, die bei Frauen häufig zu Amenorrhoe, Galaktorrhoe oder nicht-puerperaler Mastitis führt. Außerdem kommt es häufig zu einem Anstieg von 1,25-Dihydroxy-Vitamin D, dem aktiven Metaboliten von Vitamin D, der normalerweise in der Niere hydroxyliert wird. Bei Sarkoidose-Patienten kann die Hydroxylierung von Vitamin D jedoch auch außerhalb der Nieren stattfinden, nämlich in den Immunzellen in den Granulomen, die die Krankheit hervorruft. 1,25-Dihydroxy-Vitamin D ist die Hauptursache für Hyperkalzämie bei Sarkoidose und wird von Sarkoid-Granulomen überproduziert. Gamma-Interferon, das von aktivierten Lymphozyten und Makrophagen produziert wird, spielt eine wichtige Rolle bei der Synthese von 1-Alpha-25(OH)2D3. Hyperkalziurie (übermäßige Kalziumausscheidung im Urin) und Hyperkalzämie (übermäßig hohe Kalziumkonzentration im Blut) treten bei <10 % der Betroffenen auf und sind wahrscheinlich auf die erhöhte 1,25-Dihydroxy-Vitamin-D-Produktion zurückzuführen. ⓘ
Eine Schilddrüsenfehlfunktion wird in 4,2 bis 4,6 % der Fälle beobachtet. ⓘ
Eine Vergrößerung der Ohrspeicheldrüse tritt in etwa 5-10 % der Fälle auf. Eine bilaterale Beteiligung ist die Regel. Die Drüse ist normalerweise nicht empfindlich, sondern fest und glatt. Mundtrockenheit kann auftreten; andere exokrine Drüsen sind nur selten betroffen. Die Augen, ihre Drüsen oder die Ohrspeicheldrüsen sind in 20-50 % der Fälle betroffen. ⓘ
Gastrointestinal- und Urogenitalbereich
Eine symptomatische gastrointestinale (GI) Beteiligung tritt in weniger als 1 % der Fälle auf (wenn man die Leber ausschließt), und am häufigsten ist der Magen betroffen, obwohl in einem kleinen Teil der Fälle auch der Dünn- oder Dickdarm betroffen sein kann. Untersuchungen bei der Autopsie haben ergeben, dass bei weniger als 10 % der Menschen der Magen-Darm-Trakt betroffen ist. Diese Fälle würden wahrscheinlich den Morbus Crohn imitieren, bei dem es sich um eine eher den Darm betreffende granulomatöse Erkrankung handelt. Bei etwa 1 bis 3 % der Betroffenen wird bei der Autopsie eine Beteiligung der Bauchspeicheldrüse festgestellt. Eine symptomatische Nierenbeteiligung tritt nur in 0,7 % der Fälle auf, obwohl Hinweise auf eine Nierenbeteiligung bei der Autopsie in bis zu 22 % der Fälle gemeldet wurden und ausschließlich bei chronischen Erkrankungen auftreten. Bei der symptomatischen Nierenbeteiligung handelt es sich in der Regel um eine Nephrokalzinose, obwohl eine granulomatöse interstitielle Nephritis, die sich durch eine verminderte Kreatinin-Clearance und eine geringe Proteinurie auszeichnet, dicht dahinter liegt. Seltener können Nebenhoden, Hoden, Prostata, Eierstöcke, Eileiter, Gebärmutter oder die Vulva betroffen sein, wobei letztere einen Juckreiz der Vulva verursachen kann. Bei der Autopsie wurde bei etwa 5 % der Betroffenen eine Hodenbeteiligung festgestellt. Bei Männern kann die Sarkoidose zu Unfruchtbarkeit führen. ⓘ
Etwa 70 % der Menschen haben Granulome in der Leber, obwohl nur in etwa 20-30 % der Fälle Anomalien in den Leberfunktionstests festgestellt werden, die diese Tatsache widerspiegeln. Etwa 5-15 % der Patienten weisen eine Hepatomegalie auf. Nur 5-30 % der Fälle von Leberbeteiligung sind symptomatisch. In der Regel spiegeln diese Veränderungen ein cholestatisches Muster wider und umfassen erhöhte Werte der alkalischen Phosphatase (die häufigste Anomalie der Leberfunktionstests bei Sarkoidose), während Bilirubin und Aminotransferasen nur leicht erhöht sind. Gelbsucht ist selten. ⓘ
Blut
Abnormale Bluttests sind häufig und machen über 50 % der Fälle aus, sind aber nicht diagnostisch. Lymphopenie ist die häufigste Blutanomalie bei Sarkoidose. Anämie tritt bei etwa 20 % der Menschen mit Sarkoidose auf. Eine Leukopenie ist weniger häufig und tritt in noch weniger Fällen auf, ist aber selten schwerwiegend. Thrombozytopenie und hämolytische Anämie sind eher selten. Bei fehlender Splenomegalie kann die Leukopenie auf eine Knochenmarksbeteiligung hindeuten, der häufigste Mechanismus ist jedoch eine Umverteilung von T-Zellen im Blut zu den Krankheitsherden. Weitere unspezifische Befunde sind Monozytose, die in der Mehrzahl der Sarkoidosefälle auftritt, sowie erhöhte Leberenzyme oder alkalische Phosphatase. Menschen mit Sarkoidose haben häufig immunologische Anomalien wie Allergien gegen Testantigene wie Candida oder gereinigte Proteinderivate. Die polyklonale Hypergammaglobulinämie ist ebenfalls eine recht häufige immunologische Anomalie bei Sarkoidose. ⓘ
Lymphadenopathie (geschwollene Drüsen) ist bei Sarkoidose häufig und tritt in 15 % der Fälle auf. Intrathorakale Knoten sind in 75 bis 90 % aller Fälle vergrößert; in der Regel betrifft dies die Hilusknoten, aber auch die paratrachealen Knoten sind häufig betroffen. Die periphere Lymphadenopathie ist sehr häufig, vor allem die Hals- (die häufigste Kopf- und Halsmanifestation der Krankheit), Axillar-, Epitrochlear- und Leistenknoten sind betroffen. In etwa 75 % der Fälle ist die Milz mikroskopisch befallen, obwohl nur in etwa 5-10 % der Fälle eine Splenomegalie auftritt. ⓘ
Knochen, Gelenke und Muskeln
Die Sarkoidose kann Gelenke, Knochen und Muskeln betreffen. Dies führt zu einer Vielzahl von Muskel-Skelett-Beschwerden, die über unterschiedliche Mechanismen wirken. Etwa 5-15 % der Fälle betreffen die Knochen, Gelenke oder Muskeln. ⓘ
Arthritische Syndrome können als akut oder chronisch kategorisiert werden. Sarkoidosepatienten mit akuter Arthritis haben häufig auch eine bilaterale hiläre Lymphadenopathie und ein Erythema nodosum. Diese drei assoziierten Syndrome treten beim Löfgren-Syndrom häufig gemeinsam auf. Die Arthritis-Symptome des Löfgren-Syndroms treten am häufigsten an den Knöcheln auf, gefolgt von den Knien, Handgelenken, Ellenbogen und Zehengrundgelenken. In der Regel liegt keine echte Arthritis vor, sondern eine Periarthritis, die sich als Schwellung des Weichteilgewebes um die Gelenke herum zeigt und mit Hilfe von Ultraschalluntersuchungen festgestellt werden kann. Diese Gelenksymptome treten in der Regel vor oder gleichzeitig mit der Entwicklung des Erythema nodosum auf. Selbst wenn kein Erythema nodosum vorliegt, kann die Kombination aus hilärer Lymphadenopathie und Knöchelperiarthritis als eine Variante des Löfgren-Syndroms angesehen werden. Bei etwa einem Drittel der Patienten mit akuter Sarkoidarthritis tritt auch eine Enthesitis auf, die hauptsächlich die Achillessehne und die Fersen betrifft. Weichteilschwellungen an den Knöcheln können auffällig sein, und die Biopsie dieser Weichteile zeigt keine Granulome, aber eine Pannikulitis ähnlich dem Erythema nodosum. ⓘ
Die chronische Sarkoidarthritis tritt in der Regel im Zusammenhang mit einer diffuseren Organbeteiligung auf. Bei der chronischen Form können Knöchel, Knie, Handgelenke, Ellenbogen und Hände betroffen sein, und oft zeigt sich ein polyartikuläres Muster. Es kann auch eine Daktylitis ähnlich wie bei der Psoriasis-Arthritis auftreten, die mit Schmerzen, Schwellungen, überlagernden Hautrötungen und darunter liegenden knöchernen Veränderungen verbunden ist. Die Entwicklung einer Jaccoud-Arthropathie (eine nicht-erosive Deformität) ist sehr selten. ⓘ
Eine Knochenbeteiligung bei Sarkoidose wurde in 1-13 % der Fälle berichtet. Am häufigsten sind Hände und Füße betroffen, während die Wirbelsäule seltener betroffen ist. Bei der Hälfte der Patienten mit knöchernen Läsionen treten Schmerzen und Steifheit auf, während die andere Hälfte asymptomatisch bleibt. Eine Periostitis ist bei der Sarkoidose selten und kann sich am Oberschenkelknochen zeigen. ⓘ
Ursache
Die genaue Ursache der Sarkoidose ist nicht bekannt. Die derzeitige Arbeitshypothese lautet, dass die Sarkoidose bei genetisch anfälligen Personen durch eine veränderte Immunreaktion nach Exposition gegenüber einem umweltbedingten, beruflichen oder infektiösen Erreger verursacht wird. In einigen Fällen kann eine Behandlung mit Tumor-Nekrose-Faktor (TNF)-Inhibitoren wie Etanercept die Ursache sein. ⓘ
Genetik
Die Vererbbarkeit der Sarkoidose variiert je nach ethnischer Zugehörigkeit. Etwa 20 % der Afroamerikaner, die an Sarkoidose erkrankt sind, haben ein Familienmitglied mit der Krankheit, während die gleiche Zahl für europäische Amerikaner bei etwa 5 % liegt. Darüber hinaus haben bei Afroamerikanern, die offenbar schwerer und chronischer erkranken, Geschwister und Eltern von Sarkoidosefällen ein etwa 2,5-fach erhöhtes Risiko, die Krankheit zu entwickeln. Bei schwedischen Personen wurde eine Heritabilität von 39 % festgestellt. Wenn in dieser Gruppe ein Familienmitglied ersten Grades erkrankt ist, hat eine Person ein vierfach höheres Risiko, ebenfalls zu erkranken. ⓘ
Bei der Untersuchung der genetischen Anfälligkeit wurden viele Kandidatengene gefunden, aber nur wenige wurden durch weitere Untersuchungen bestätigt, und es sind keine zuverlässigen genetischen Marker bekannt. Das derzeit interessanteste Kandidatengen ist BTNL2; auch mehrere HLA-DR-Risikoallele werden untersucht. Bei persistierender Sarkoidose ist der HLA-Haplotyp HLA-B7-DR15 entweder an der Krankheit beteiligt oder ein anderes Gen zwischen diesen beiden Loci ist assoziiert. Bei nicht persistierender Erkrankung besteht ein starker genetischer Zusammenhang mit HLA DR3-DQ2. Kardiales Sarkoid wurde mit Varianten des Tumor-Nekrose-Faktors alpha (TNFA) in Verbindung gebracht. ⓘ
Infektiöse Erreger
Mehrere Infektionserreger scheinen signifikant mit Sarkoidose assoziiert zu sein, aber keiner der bekannten Zusammenhänge ist spezifisch genug, um auf eine direkte ursächliche Rolle hinzuweisen. Zu den wichtigsten in Frage kommenden Infektionserregern gehören Mykobakterien, Pilze, Borrelien und Rickettsien. Eine Meta-Analyse, die die Rolle von Mykobakterien bei der Sarkoidose untersuchte, ergab, dass sie in 26,4 % der Fälle vorkommen, aber es wurde auch eine mögliche Publikationsverzerrung festgestellt, so dass die Ergebnisse weiter bestätigt werden müssen. Die Katalase-Peroxidase von Mycobacterium tuberculosis wurde als möglicher Antigen-Katalysator der Sarkoidose identifiziert. Es wurde auch über eine Übertragung der Krankheit durch Organtransplantationen berichtet. Eine große epidemiologische Studie fand wenig Hinweise darauf, dass Infektionskrankheiten, die Jahre vor der Sarkoidose-Diagnose aufgetreten sind, ein messbares Risiko für die Sarkoidose-Diagnose in der Zukunft darstellen könnten. ⓘ
Autoimmunerkrankungen
Eine Assoziation mit Autoimmunerkrankungen ist häufig beobachtet worden. Der genaue Mechanismus dieses Zusammenhangs ist nicht bekannt, aber es gibt Anhaltspunkte für die Hypothese, dass dies eine Folge der Th1-Lymphokinprävalenz ist. Tests der verzögerten kutanen Hypersensitivität wurden zur Messung der Progression verwendet. ⓘ
Pathophysiologie
Die granulomatöse Entzündung ist in erster Linie durch die Ansammlung von Makrophagen und aktivierten T-Lymphozyten gekennzeichnet, mit einer erhöhten Produktion der wichtigsten Entzündungsmediatoren, Tumornekrosefaktor alpha (TNF), Interferon gamma, Interleukin 2 (IL-2), IL-8, IL-10, IL-12, IL-18, IL-23 und transformierender Wachstumsfaktor beta (TGF-β), die auf eine T-Helferzellen-vermittelte Immunantwort hinweisen. Die Sarkoidose hat paradoxe Auswirkungen auf die Entzündungsprozesse; sie ist durch eine erhöhte Makrophagen- und CD4-Helfer-T-Zell-Aktivierung gekennzeichnet, was zu einer beschleunigten Entzündung führt, während die Immunantwort auf Antigenherausforderungen wie Tuberkulin unterdrückt ist. Dieser paradoxe Zustand der gleichzeitigen Hyper- und Hypoaktivität deutet auf einen Zustand der Anergie hin. Die Anergie könnte auch für das erhöhte Infektions- und Krebsrisiko verantwortlich sein. Die regulatorischen T-Lymphozyten in der Peripherie von Sarkoidgranulomen scheinen die IL-2-Sekretion zu unterdrücken, die, so die Hypothese, den Zustand der Anergie verursacht, indem sie antigenspezifische Gedächtnisreaktionen verhindert. ⓘ
Es wird zwar allgemein angenommen, dass TNF eine wichtige Rolle bei der Bildung von Granulomen spielt (dies wird auch durch die Feststellung gestützt, dass in Tiermodellen für die Bildung von mykobakteriellen Granulomen die Hemmung der TNF- oder IFN-γ-Produktion die Granulombildung hemmt), aber Sarkoidose kann sich auch bei Patienten entwickeln, die mit TNF-Antagonisten wie Etanercept behandelt werden. Auch B-Zellen spielen wahrscheinlich eine Rolle in der Pathophysiologie der Sarkoidose. Die Serumspiegel der löslichen humanen Leukozytenantigene (HLA) der Klasse I und des Angiotensin-Converting-Enzyms (ACE) sind bei Menschen mit Sarkoidose erhöht. Auch das Verhältnis von CD4/CD8-T-Zellen in der bronchoalveolären Lavage ist bei Menschen mit pulmonaler Sarkoidose in der Regel höher (in der Regel >3,5), obwohl es in einigen Fällen normal oder sogar abnormal niedrig sein kann. Es hat sich gezeigt, dass der ACE-Serumspiegel in der Regel mit der Gesamtbelastung durch Granulome korreliert. ⓘ
Fälle von Sarkoidose wurden auch im Rahmen des HIV-Immunrekonstitutionssyndroms gemeldet, d. h., wenn Menschen eine HIV-Behandlung erhalten, erholt sich ihr Immunsystem und beginnt, die Antigene opportunistischer Infektionen anzugreifen, die es sich vor dieser Erholung eingefangen hat, und die daraus resultierende Immunreaktion beginnt, gesundes Gewebe zu schädigen. ⓘ
Histopathologie
Die Sarkoidose ist durch die Bildung von nicht-nekrotisierenden ("nicht-verkäsenden") Granulomen in verschiedenen Organen und Geweben gekennzeichnet. Riesenzellen, insbesondere Langhans-Riesenzellen, sind bei Sarkoidose häufig anzutreffen. Schaumann-Körperchen, die bei Sarkoidose auftreten, sind Kalzium- und Proteineinschlüsse innerhalb von Riesenzellen als Teil eines Granuloms. Asteroide Körper können bei Sarkoidose vorkommen. Hamazaki-Wesenberg-Körper können in Lymphknoten und seltener in Lungenbiopsien mit Sarkoidose gesehen werden und sind Einschlusskörper von Lysosomen mit Protein, Glykoprotein und Eisen. ⓘ

Sarkoidose in einem Lymphknoten
- Image:Asteroid body intermed mag.jpg
Lungensarkoidose mit Granulomen mit Langhans-Riesenzellen und asteroiden Körpern
- Image:Sarcoidosis - Schaumann body 2.jpg
Schaumann-Körper bei Sarkoidose
- Image:Sarcoidosis - Asteroid body (6152059052).jpg
Asteroidkörper bei Sarkoidose
- Image:Sarcoidosis - Hamazaki-Wesenberg (H-W) bodies- Lymph node (6134890353).jpg
Hamazaki-Wesenberg-Körper bei Sarkoidose im Lymphknoten ⓘ
_lymph_node_biopsy.jpg)
Diagnose

Die Diagnose der Sarkoidose ist eine Ausschlussdiagnose, da es keinen spezifischen Test für diese Erkrankung gibt. Zum Ausschluss einer Sarkoidose bei einem Fall mit pulmonalen Symptomen können eine Röntgenaufnahme des Brustkorbs, eine CT-Untersuchung des Brustkorbs, eine PET-Untersuchung, eine CT-gesteuerte Biopsie, eine Mediastinoskopie, eine offene Lungenbiopsie, eine Bronchoskopie mit Biopsie, ein endobronchialer Ultraschall und ein endoskopischer Ultraschall mit Feinnadelaspiration der mediastinalen Lymphknoten (EBUS FNA) durchgeführt werden. Das Gewebe aus der Biopsie der Lymphknoten wird sowohl einer Durchflusszytometrie unterzogen, um Krebs auszuschließen, als auch speziellen Färbungen (Färbung mit säurefesten Bazillen und Gömöri-Methenamin-Silberfärbung), um Mikroorganismen und Pilze auszuschließen. ⓘ
Zu den Serummarkern der Sarkoidose gehören: Serum-Amyloid A, löslicher Interleukin-2-Rezeptor, Lysozym, Angiotensin-Converting Enzyme und das Glykoprotein KL-6. Die Blutspiegel des Angiotensin-konvertierenden Enzyms werden bei der Überwachung der Sarkoidose verwendet. In einer bronchoalveolären Lavage kann ein erhöhtes (von mindestens 3,5) CD4/CD8-T-Zell-Verhältnis nachgewiesen werden, was ein Hinweis (aber kein Beweis) für eine Lungensarkoidose ist. In mindestens einer Studie korrelierte das induzierte CD4/CD8-Verhältnis im Sputum und die TNF-Konzentration mit den Werten in der Lavageflüssigkeit. Eine Sarkoidose-ähnliche Lungenerkrankung, die granulomatös-lymphozytäre interstitielle Lungenerkrankung, kann bei Patienten mit allgemeinem variablen Immundefekt (CVID) auftreten, weshalb zum Ausschluss von CVID die Serumantikörperspiegel gemessen werden sollten. ⓘ
Zu den Differentialdiagnosen gehören metastatische Erkrankungen, Lymphome, septische Embolien, rheumatische Knoten, Granulomatose mit Polyangiitis, Varizelleninfektion, Tuberkulose und atypische Infektionen wie Mycobacterium avium complex, Cytomegalovirus und Kryptokokken. Die Sarkoidose wird am häufigsten mit neoplastischen Erkrankungen, wie z. B. Lymphomen, oder mit Erkrankungen verwechselt, die ebenfalls durch einen granulomatösen Entzündungsprozess mit mononukleären Zellen gekennzeichnet sind, wie z. B. die mykobakteriellen und pilzbedingten Erkrankungen. ⓘ
Die Veränderungen im Röntgenbild des Brustkorbs werden in vier Stadien eingeteilt:
- Bihiläre Lymphadenopathie
- Bihiläre Lymphadenopathie und retikulonoduläre Infiltrate
- bilaterale pulmonale Infiltrate
- Fibrozystische Sarkoidose, typischerweise mit Einziehung des Hilus nach oben, zystischen und bullösen Veränderungen
Obwohl Menschen mit Röntgenbildern im Stadium 1 dazu neigen, die akute oder subakute, reversible Form der Krankheit zu haben, haben diejenigen mit den Stadien 2 und 3 oft die chronische, fortschreitende Krankheit; diese Muster stellen keine aufeinanderfolgenden "Stadien" der Sarkoidose dar. Abgesehen von epidemiologischen Zwecken ist diese Kategorisierung daher hauptsächlich von historischem Interesse. ⓘ
Bei Sarkoidose, die in der kaukasischen Bevölkerung auftritt, sind hiläre Adenopathie und Erythema nodosum die häufigsten Anfangssymptome. In dieser Bevölkerungsgruppe ist eine Biopsie des Gastrocnemius-Muskels ein nützliches Instrument für die korrekte Diagnose. Das Vorhandensein eines nicht kaseinierenden epithelioiden Granuloms in einer Gastrocnemius-Probe ist der definitive Beweis für eine Sarkoidose, da andere tuberkuloide und pilzbedingte Erkrankungen in diesem Muskel histologisch äußerst selten auftreten. ⓘ
Die kardiale Magnetresonanztomographie (CMR) ist ein Verfahren zur Diagnose der kardialen Sarkoidose. Sie hat eine Spezifität von 78 % für die Diagnose der kardialen Sarkoidose. Mit der T2-gewichteten Bildgebung kann eine akute Entzündung nachgewiesen werden. Mit dem späten Gadoliniumkontrast (LGE) können Fibrose oder Narben nachgewiesen werden. Läsionen am Subperikard und eine Anreicherung in der Mitte des Septum basale oder der inferolateralen Wand sind ein starker Hinweis auf Sarkoidose. Mit der MRT können auch die Wirksamkeit der Behandlung mit Kortikosteroiden und die Prognose der kardialen Sarkoidose verfolgt werden. ⓘ
Mit einem PET-Scan lässt sich die Krankheitsaktivität quantifizieren, was mit der CMR nicht möglich ist. ⓘ
Hiläre Adenopathie, insbesondere auf der linken Seite (AP CXR)

Hiläre Adenopathie vor allem links (laterale CXR)

Adenopathie der Hilusorgane, insbesondere links (koronales CT)

Adenopathie der Hilusorgane, insbesondere links (transversales CT) ⓘ




Klassifizierung
Die Sarkoidose kann in die folgenden Typen unterteilt werden:
- Annuläre Sarkoidose
- Erythrodermische Sarkoidose
- Ichthyosiforme Sarkoidose
- Hypopigmentierte Sarkoidose
- Löfgren-Syndrom
- Lupus pernio
- Morpheaforme Sarkoidose
- Mukosale Sarkoidose
- Neurosarkoidose
- Papuläres Sarkoid
- Narbensarkoid
- Subkutane Sarkoidose
- Systemische Sarkoidose
- Ulzerative Sarkoidose ⓘ
Behandlung
Eine kausale Therapie existiert bei der Sarkoidose nicht. Eine Therapie ist sinnvoll, wenn Beschwerden oder Komplikationen bestehen. Da bis zu 60 % der Sarkoidosen eine Spontanremission haben, kann man zunächst engmaschig beobachten und versuchen, die unterschiedlichen Beschwerden symptomatisch zu lindern. Hingegen bei symptomatischem Organbefall wird eine Cortisontherapie durchgeführt, zum Beispiel bei funktionellen Einschränkungen der Lunge, davon insbesondere im Stadium III oder bei Hyperkalzämie (unter anderem bei Knochenmarksbefall). Die Dosierung beträgt oft 20–80 mg/Tag Prednisolon, nach einer Zeit kann versucht werden, zu reduzieren und dann auszuschleichen. Steroidbedingte Nebenwirkungen sollten möglichst gering gehalten werden. In manchen Fällen wird auch Methotrexat, meist in einer Dosierung von 10–15 mg/Woche, verwendet, um die Cortisondosis zu reduzieren. In der Langzeittherapie können auch Immunsuppressiva wie Azathioprin und Chloroquin verwendet werden. Alle Therapien müssen engmaschig ärztlich überwacht werden. ⓘ
Beim Löfgren-Syndrom und in akuten Schüben werden statt (oder zusätzlich zu) Corticoiden auch Acetylsalicylsäure, Ibuprofen oder Diclofenac, gegebenenfalls auch zusätzlich Schmerzmittel eingesetzt. Wenn die Hauterscheinungen bei der Sarkoidose im Vordergrund stehen (kutane Sarkoidose), kann eine Therapie mit Tetracyclinen oder Allopurinol versucht werden. Der Wirkmechanismus von Allopurinol ist hierbei ungeklärt. ⓘ
Der Zeitpunkt des Beginns der Therapie mit Corticosteroiden bei Sarkoidose ist nicht unumstritten, da diese Therapie die Symptome nur unterdrückt und bei Beendigung der Therapie wieder auftreten lassen könnte. Eine Untersuchung zur Kortisontherapie kam zu dem Ergebnis, dass die Behandlung mit Cortison die Rückfallquote wesentlich erhöhe. Als mögliche Ursache wird zum einen die Cortisonbehandlung an sich genannt, alternativ könnte es auch einfach darauf zurückzuführen sein, dass überwiegend schwerere Krankheitsverläufe mit Cortison behandelt wurden. Kleine Studien geben Hinweise auf Wirkung des TNF-α-Blockers Infliximab wie auch bei anderen Krankheitsbildern aus dem rheumatischen Formenkreis. Möglicherweise wird aber damit die Sterblichkeit erhöht. ⓘ
Eine begleitende psychotherapeutische Behandlung kann sich günstig auf den Krankheitsverlauf auswirken. ⓘ
Die Behandlungen der Sarkoidose sind je nach Patient sehr unterschiedlich. Mindestens die Hälfte der Patienten benötigt keine systemische Therapie. Die meisten (>75 %) benötigen nur eine symptomatische Behandlung mit nichtsteroidalen Antirheumatika (NSAIDs) wie Ibuprofen oder Aspirin. Bei Patienten mit Lungensymptomen wird die aktive pulmonale Sarkoidose in der Regel zwei bis drei Monate lang ohne Therapie beobachtet; wenn die Entzündung nicht spontan abklingt, wird eine Therapie eingeleitet. ⓘ
Zu den wichtigsten Kategorien medikamentöser Interventionen gehören Glukokortikoide, Antimetaboliten und biologische Wirkstoffe, insbesondere monoklonale Antikörper gegen den Tumornekrosefaktor. Zu den in der Erforschung befindlichen Behandlungen gehören spezifische Antibiotika-Kombinationen und mesenchymale Stammzellen. Wenn eine medikamentöse Behandlung angezeigt ist, wird häufig ein schrittweises Vorgehen angewandt, um Alternativen in der Reihenfolge der zunehmenden Nebenwirkungen zu prüfen und potenziell toxische Wirkungen zu überwachen. ⓘ
Kortikosteroide, am häufigsten Prednison oder Prednisolon, sind seit vielen Jahren die Standardbehandlung. Bei einigen Menschen kann diese Behandlung den Krankheitsverlauf verlangsamen oder umkehren, andere sprechen jedoch nicht auf eine Steroidtherapie an. Der Einsatz von Kortikosteroiden bei leichter Erkrankung ist umstritten, da die Krankheit in vielen Fällen spontan zurückgeht. ⓘ
Antimetaboliten
Antimetabolite, die auch als steroidsparende Mittel bezeichnet werden, wie Azathioprin, Methotrexat, Mycophenolsäure und Leflunomid, werden häufig als Alternative zu Kortikosteroiden eingesetzt. Von diesen Mitteln wird Methotrexat am häufigsten eingesetzt und untersucht. Methotrexat gilt als Erstlinientherapie bei Neurosarkoidose, oft in Verbindung mit Kortikosteroiden. Eine Langzeitbehandlung mit Methotrexat wird bei etwa 10 % der Patienten mit Leberschäden in Verbindung gebracht und kann daher bei Patienten mit Leberbeteiligung ein erhebliches Problem darstellen und erfordert eine regelmäßige Überwachung der Leberfunktionstests. Methotrexat kann auch zu einer Lungentoxizität (Lungenschädigung) führen, obwohl dies eher selten vorkommt und eher die durch Sarkoidose verursachte Leukopenie verkomplizieren kann. Aufgrund dieser Sicherheitsbedenken wird häufig empfohlen, Methotrexat mit Folsäure zu kombinieren, um Toxizität zu vermeiden. Auch die Behandlung mit Azathioprin kann zu Leberschäden führen. Allerdings scheint das Infektionsrisiko bei Patienten, die mit Methotrexat anstelle von Azathioprin behandelt werden, um etwa 40 % geringer zu sein. Leflunomid wird als Ersatz für Methotrexat verwendet, möglicherweise aufgrund seiner angeblich geringeren Lungentoxizität. Mycophenolsäure wurde erfolgreich bei der uvealen Sarkoidose, der Neurosarkoidose (insbesondere bei der ZNS-Sarkoidose; geringfügig wirksam bei der Sarkoidose-Myopathie) und der pulmonalen Sarkoidose eingesetzt. ⓘ
Immunsuppressiva
Da die Granulome durch Ansammlungen von Zellen des Immunsystems, insbesondere von T-Zellen, verursacht werden, wurden einige Erfolge mit Immunsuppressiva (wie Cyclophosphamid, Cladribin, Chlorambucil und Cyclosporin), immunmodulatorischen Mitteln (Pentoxifyllin und Thalidomid) und Medikamenten gegen Tumornekrosefaktoren (wie Infliximab, Etanercept, Golimumab und Adalimumab) erzielt. ⓘ
In einer klinischen Studie konnte Cyclosporin, das zusätzlich zur Prednisonbehandlung verabreicht wurde, bei Patienten mit pulmonaler Sarkoidose keinen signifikanten Vorteil gegenüber Prednison allein nachweisen, obwohl es Hinweise auf eine erhöhte Toxizität durch die zusätzliche Verabreichung von Cyclosporin zur Steroidbehandlung gab, darunter Infektionen, maligne Erkrankungen (Krebserkrankungen), Bluthochdruck und Nierenfunktionsstörungen. Auch Chlorambucil und Cyclophosphamid werden bei der Behandlung der Sarkoidose wegen ihrer hohen Toxizität, insbesondere wegen ihres Potenzials, bösartige Erkrankungen auszulösen, nur selten eingesetzt. Infliximab wurde in klinischen Studien in einer Reihe von Fällen erfolgreich zur Behandlung der pulmonalen Sarkoidose eingesetzt. Etanercept hingegen hat in einigen klinischen Studien keine signifikante Wirksamkeit bei Patienten mit Aderhautsarkoidose gezeigt. Auch Golimumab hat bei Patienten mit pulmonaler Sarkoidose keinen Nutzen gezeigt. In einer klinischen Studie mit Adalimumab wurde bei etwa der Hälfte der Probanden ein Ansprechen auf die Behandlung festgestellt, was mit der Wirkung von Infliximab vergleichbar ist; da Adalimumab jedoch ein besseres Verträglichkeitsprofil aufweist, wird es möglicherweise gegenüber Infliximab bevorzugt. ⓘ
Spezifische Organbehandlungen
Ursodeoxycholsäure wurde erfolgreich zur Behandlung von Fällen mit Leberbeteiligung eingesetzt. Auch Thalidomid wurde in einer klinischen Studie erfolgreich zur Behandlung des therapieresistenten Lupus pernio eingesetzt, was möglicherweise auf seine Anti-TNF-Wirkung zurückzuführen ist, obwohl es in einer klinischen Studie zur Lungensarkoidose keine Wirksamkeit zeigte. Die kutane Erkrankung kann mit Malariamitteln (wie Chloroquin und Hydroxychloroquin) und dem Tetrazyklin-Antibiotikum Minozyklin erfolgreich behandelt werden. Antimalariamittel haben sich auch bei der Behandlung der Sarkoidose-bedingten Hyperkalzämie und der Neurosarkoidose als wirksam erwiesen. Der langfristige Einsatz von Antimalariamitteln ist jedoch durch ihr Potenzial, irreversible Erblindung zu verursachen, eingeschränkt, weshalb regelmäßige augenärztliche Untersuchungen erforderlich sind. Diese Toxizität ist bei Hydroxychloroquin in der Regel weniger problematisch als bei Chloroquin, obwohl Hydroxychloroquin die Glukosehomöostase stören kann. ⓘ
Kürzlich wurden selektive Phosphodiesterase 4 (PDE4)-Hemmer wie Apremilast (ein Thalidomid-Derivat), Roflumilast und der weniger subtypselektive PDE4-Hemmer Pentoxifyllin als Behandlung der Sarkoidose erprobt, wobei Apremilast in einer kleinen offenen Studie bei kutaner Sarkoidose erfolgreiche Ergebnisse erzielte. Pentoxifyllin wurde erfolgreich zur Behandlung der akuten Erkrankung eingesetzt, obwohl sein Einsatz durch seine gastrointestinale Toxizität (meist Übelkeit, Erbrechen und Durchfall) stark eingeschränkt ist. Fallberichte haben die Wirksamkeit von Rituximab, einem monoklonalen Anti-CD20-Antikörper, bestätigt, und eine klinische Studie zur Untersuchung von Atorvastatin als Behandlung der Sarkoidose ist im Gange. Es wurde berichtet, dass ACE-Hemmer bei kutaner Sarkoidose eine Remission und bei pulmonaler Sarkoidose eine Verbesserung bewirken, einschließlich einer Verbesserung der Lungenfunktion, eines Remodellings des Lungenparenchyms und einer Verhinderung der Lungenfibrose in separaten Fallserien". Es wurde festgestellt, dass Nikotinpflaster bei Sarkoidosepatienten entzündungshemmende Wirkungen haben, ob sie jedoch krankheitsverändernde Wirkungen haben, muss weiter untersucht werden. Eine antimykobakterielle Behandlung (Medikamente, die Mykobakterien, die Erreger von Tuberkulose und Lepra, abtöten) hat sich in einer klinischen Studie ebenfalls als wirksam bei der Behandlung der chronisch kutanen (d. h. die Haut betreffenden) Sarkoidose erwiesen. Quercetin wurde auch als Mittel zur Behandlung der Lungensarkoidose erprobt, wobei in einer kleinen Studie erste Erfolge erzielt wurden. ⓘ
Aufgrund ihrer Seltenheit ist die Behandlung der Sarkoidose der männlichen Geschlechtsorgane umstritten. Da die Differentialdiagnose auch Hodenkrebs einschließt, empfehlen einige eine Orchiektomie, selbst wenn Anzeichen für eine Sarkoidose in anderen Organen vorhanden sind. Bei dem neueren Ansatz wird eine Biopsie von Hoden und Nebenhoden und die Resektion der größten Läsion vorgeschlagen. ⓘ
Symptome
Menschen mit Sarkoidose können eine Reihe von Symptomen haben, die nicht mit objektiven körperlichen Anzeichen der Krankheit übereinstimmen, aber dennoch die Lebensqualität beeinträchtigen. ⓘ
Physiotherapie, Rehabilitation und Beratung können dazu beitragen, eine Dekonditionierung zu vermeiden und die soziale Teilhabe, das psychische Wohlbefinden und das Aktivitätsniveau zu verbessern. Wichtige Aspekte sind die Vermeidung von Belastungsintoleranz und Muskelschwäche. ⓘ
Es hat sich gezeigt, dass körperliches Training mit geringer oder mäßiger Intensität die Müdigkeit, das psychische Wohlbefinden und die körperliche Funktionsfähigkeit bei Sarkoidosepatienten ohne negative Auswirkungen verbessert. Inspiratorisches Muskeltraining hat auch bei Personen mit Sarkoidose im Frühstadium die Wahrnehmung schwerer Ermüdung verringert und die funktionelle und maximale Belastbarkeit sowie die Atemmuskelkraft verbessert. Dauer, Häufigkeit und Intensität des Trainings müssen auf Beeinträchtigungen wie Gelenkschmerzen, Muskelschmerzen und Müdigkeit abgestimmt werden. ⓘ
Neurostimulanzien wie Methylphenidat und Modafinil haben sich als Ergänzung zur Behandlung der Sarkoidose-Müdigkeit als wirksam erwiesen. ⓘ
Die Behandlung symptomatischer neuropathischer Schmerzen bei Sarkoidosepatienten ist ähnlich wie bei anderen Ursachen und umfasst Antidepressiva, Antikonvulsiva und Opioide mit verlängerter Wirkstofffreisetzung. Allerdings erfahren nur 30 bis 60 % der Patienten eine begrenzte Schmerzlinderung. ⓘ
Prognose
Die Krankheit kann spontan remittieren oder chronisch werden, mit Exazerbationen und Remissionen. In einigen Fällen kann sie zu Lungenfibrose und Tod führen. In gutartigen Fällen kann die Remission ohne Behandlung innerhalb von 24 bis 36 Monaten eintreten, doch sind regelmäßige Nachuntersuchungen erforderlich. Einige Fälle können jedoch mehrere Jahrzehnte andauern. Zwei Drittel der Erkrankten erreichen innerhalb von 10 Jahren nach der Diagnose eine Remission. Wenn das Herz betroffen ist, ist die Prognose im Allgemeinen weniger günstig, obwohl Kortikosteroide bei der Verbesserung der AV-Überleitung wirksam zu sein scheinen. Die Prognose ist bei Afroamerikanern tendenziell ungünstiger als bei weißen Amerikanern. In einer schwedischen bevölkerungsbasierten Analyse wies die Mehrheit der Fälle, die zum Zeitpunkt der Diagnose keine schwere Erkrankung aufwiesen, eine mit der Allgemeinbevölkerung vergleichbare Sterblichkeit auf. Das Risiko eines vorzeitigen Todes war bei einer kleineren Gruppe von Patienten, die zum Zeitpunkt der Diagnose eine schwere Erkrankung aufwiesen, im Vergleich zur Allgemeinbevölkerung deutlich (um das 2,3-fache) erhöht. Schwere Infektionen, die im Verlauf der Krankheit manchmal mehrfach auftreten, und Herzversagen könnten zu dem höheren Risiko eines frühen Todes bei einigen Sarkoidosepatienten beitragen. ⓘ
Einige Studien aus den 1990er Jahren deuten darauf hin, dass Menschen mit Sarkoidose offenbar ein deutlich erhöhtes Risiko für Krebs haben, insbesondere für Lungenkrebs, Lymphome und Krebs in anderen Organen, die bekanntermaßen bei Sarkoidose betroffen sind. Beim Sarkoidose-Lymphom-Syndrom folgt auf die Sarkoidose die Entwicklung einer lymphoproliferativen Störung wie das Non-Hodgkin-Lymphom. Dies kann auf die zugrundeliegenden immunologischen Anomalien zurückgeführt werden, die während des Krankheitsprozesses der Sarkoidose auftreten. Sarkoidose kann auch nach einer Krebserkrankung oder gleichzeitig mit einer Krebserkrankung auftreten. Es gibt Berichte über Haarzellenleukämie, akute myeloische Leukämie und akute myeloblastische Leukämie in Verbindung mit Sarkoidose. Manchmal kann eine Sarkoidose, auch wenn sie unbehandelt ist, durch opportunistische Infektionen kompliziert werden, obwohl diese selten sind. ⓘ
Epidemiologie
Die Sarkoidose betrifft am häufigsten junge Erwachsene beiderlei Geschlechts, obwohl in Studien mehr Fälle bei Frauen gemeldet wurden. Die Inzidenz ist bei Personen unter 40 Jahren am höchsten und erreicht ihren Höhepunkt in der Altersgruppe von 20 bis 29 Jahren; ein zweiter Höhepunkt wird bei Frauen über 50 Jahren beobachtet. ⓘ
Sarkoidose tritt weltweit bei allen Rassen auf, mit einer durchschnittlichen Inzidenz von 16,5 pro 100.000 bei Männern und 19 pro 100.000 bei Frauen. Die Krankheit ist in den nordeuropäischen Ländern am weitesten verbreitet, und die höchste jährliche Inzidenz von 60 pro 100 000 ist in Schweden und Island zu verzeichnen. Im Vereinigten Königreich liegt die Prävalenz bei 16 von 100.000. In den Vereinigten Staaten ist die Sarkoidose bei Menschen afrikanischer Abstammung häufiger als bei Kaukasiern, wobei die jährliche Inzidenz mit 35,5 bzw. 10,9 pro 100 000 angegeben wird. Sarkoidose wird in Südamerika, Spanien, Indien, Kanada und auf den Philippinen seltener gemeldet. Möglicherweise besteht bei Menschen mit Zöliakie eine höhere Anfälligkeit für Sarkoidose. Es wurde ein Zusammenhang zwischen den beiden Erkrankungen vermutet. ⓘ
Es wurde auch eine saisonale Häufung von Sarkoidose-Betroffenen beobachtet. In Griechenland werden jedes Jahr etwa 70 % der Diagnosen zwischen März und Mai gestellt, in Spanien etwa 50 % der Diagnosen zwischen April und Juni, und in Japan wird die Sarkoidose meist im Juni und Juli diagnostiziert. ⓘ
Die unterschiedliche Inzidenz in der Welt kann zumindest teilweise darauf zurückzuführen sein, dass es in bestimmten Regionen der Welt keine Screening-Programme gibt und dass andere granulomatöse Erkrankungen wie Tuberkulose die Diagnose der Sarkoidose überschatten, wenn sie weit verbreitet sind. Möglicherweise gibt es auch Unterschiede im Schweregrad der Krankheit zwischen Menschen verschiedener Ethnien. Mehrere Studien deuten darauf hin, dass die Sarkoidose bei Menschen afrikanischer Herkunft schwerer und disseminierter verläuft als bei Kaukasiern, bei denen die Krankheit eher asymptomatisch ist. Die Manifestation scheint je nach Rasse und Geschlecht leicht unterschiedlich zu sein. Erythema nodosum tritt bei Männern weitaus häufiger auf als bei Frauen und bei Kaukasiern häufiger als bei anderen Rassen. Bei Japanern sind die Augen und das Herz häufiger betroffen als bei anderen Rassen. ⓘ
Sie tritt häufiger in bestimmten Berufen auf, insbesondere bei Feuerwehrleuten, Pädagogen, Militärangehörigen, Personen, die in Industrien arbeiten, in denen Pestizide verwendet werden, bei der Polizei und bei medizinischem Personal. Im Jahr nach den Anschlägen vom 11. September stieg die Sarkoidose-Inzidenzrate um das Vierfache (auf 86 Fälle pro 100 000). ⓘ
Geschichte
Als Erstbeschreiber der Sarkoidose (weitere Synonyme: Lymphogranuloma(tosis) benigna, Boeck-Besnier-Schaumannsche Krankheit, Lungentuberkuloid, benignes Miliarlupoid, multiples benignes Sarkoid, epitheloidzellige Reticuloendotheliose) kann Jonathan Hutchinson (1828–1913) angesehen werden. Er stellte 1863 einen an Gicht erkrankten Patienten vor, der zusätzlich Hautveränderungen aufwies und vier Jahre später an Nierenversagen verstarb. Hutchinson führte dies jedoch auf die Gicht zurück – heute vermutet man, dass der aufgrund Sarkoidose veränderte Calciumstoffwechsel die eigentliche Ursache war. Der französische Dermatologe Ernest Henri Besnier (1831–1909) beschrieb 1889 eine symmetrische Hautveränderung der Extremitäten. Sein norwegischer Kollege Cæsar Peter Møller Boeck (1845–1917) erwähnte 1899 in dem wissenschaftlichen Aufsatz multiple benign sarcoid of the skin die histologischen Hautveränderungen und stellte schon damals den Verdacht einer systemischen Erkrankung. Die Hautveränderungen sind seitdem als Boecksche Sarkoidose bekannt. ⓘ
Der dänische Ophthalmologe Christian Frederick Heerfordt (1871–1953) beschrieb 1909 eine fieberhafte Entzündung der Bindehaut mit Nervenbeteiligung und ordnete dies aufgrund der Laborwerte einem Mumps zu. Im Jahre 1924 bestätigte der schwedische Dermatologe Jörgen Nilsen Schaumann (1879–1953) die Entdeckungen Boecks, dass es sich hierbei um eine systemische Erkrankung verschiedener Organe handelt, und bezeichnete die Sarkoidose als Lymphogranulomatosis benigna, um sie vom Hodgkin-Lymphom abzugrenzen. ⓘ
1941 beschrieb der norwegische Arzt Morten A. Kveim (1892–1966) den Kveim-Test zur Diagnostik der Sarkoidose, in den 1990er Jahren kam er allerdings außer Gebrauch. ⓘ
Der Schwede Sven Halvar Löfgren (1910–1978) beschrieb 1953 die akute Verlaufsform anhand der Symptomtrias Erythema nodosum, Arthritis und bihiläre Lymphadenopathie (beidseitiger Befall der Lymphknoten in der Lunge). Dieses oft bei jungen Menschen anzutreffende Krankheitsbild wird als Löfgren-Syndrom bezeichnet. ⓘ
Er wurde erstmals 1877 von Dr. Jonathan Hutchinson, einem Dermatologen, als eine Erkrankung beschrieben, die rote, erhabene Ausschläge im Gesicht, an den Armen und Händen verursacht. Im Jahr 1889 wurde der Begriff Lupus pernio von Dr. Ernest Besnier, einem weiteren Dermatologen, geprägt. Später, 1892, wurde die Histologie des Lupus pernio definiert. Im Jahr 1902 beschrieb eine Gruppe von drei Ärzten erstmals die Knochenbeteiligung. Zwischen 1909 und 1910 wurde erstmals die Uveitis bei Sarkoidose beschrieben, und 1915 wurde von Dr. Jörgen Nielsen Schaumann betont, dass es sich um eine systemische Erkrankung handelt. Im selben Jahr wurde auch eine Lungenbeteiligung beschrieben. 1937 wurde das Uveoparotidalfieber und 1941 das Löfgren-Syndrom erstmals beschrieben. 1958 wurde in London die erste internationale Konferenz über Sarkoidose einberufen, ebenso fand 1961 in Washington, D.C., die erste Sarkoidosekonferenz der USA statt. Sie wurde auch als Besnier-Boeck-Krankheit oder Besnier-Boeck-Schaumann-Krankheit bezeichnet. ⓘ
Etymologie
Das Wort "Sarkoidose" stammt vom griechischen [σάρκο-] sarco-, was "Fleisch" bedeutet, dem Suffix -(e)ido (vom griechischen εἶδος -eidos [im Englischen wird das anfängliche e gewöhnlich weggelassen, da der Diphthong epsilon-iota im klassischen Griechisch für ein langes "i" = englisches ee steht]), was "Art", "ähnelt" oder "wie" bedeutet, und -sis, einem im Griechischen üblichen Suffix, das "Zustand" bedeutet. Das ganze Wort bedeutet also "ein Zustand, der rohem Fleisch ähnelt". Die ersten Fälle von Sarkodose, die als neue pathologische Entität anerkannt wurden, wiesen Ende des 19. Jahrhunderts in Skandinavien Hautknötchen auf, die kutanen Sarkomen ähnelten, daher der ursprüngliche Name. ⓘ
Gesellschaft und Kultur
Die World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) ist eine Organisation von Ärzten, die sich mit der Diagnose und Behandlung von Sarkoidose und verwandten Erkrankungen befassen. Die WASOG gibt die Zeitschrift Sarcoidosis, Vasculitis and Diffuse Lung Diseases heraus. Darüber hinaus widmet sich die Stiftung für Sarkoidoseforschung (Foundation for Sarcoidosis Research, FSR) der Erforschung der Sarkoidose und ihrer möglichen Behandlung. ⓘ
Es gibt Befürchtungen, dass die Rettungskräfte des World Trade Center einem erhöhten Sarkoidoserisiko ausgesetzt sind. ⓘ
Der Komiker und Schauspieler Bernie Mac hatte Sarkoidose. Im Jahr 2005 gab er an, dass die Krankheit in Remission sei. Sein Tod am 9. August 2008 wurde durch Komplikationen einer Lungenentzündung verursacht, obwohl Macs Agent angab, dass die Sarkoidose nicht mit seiner tödlichen Lungenentzündung zusammenhing. ⓘ
Bei Karen "Duff" Duffy, MTV-Persönlichkeit und Schauspielerin, wurde 1995 eine Neurosarkoidose diagnostiziert. ⓘ
Der American-Football-Spieler Reggie White starb 2004. Lungen- und Herzsarkoidose waren mitverantwortlich für seine tödlichen Herzrhythmusstörungen. ⓘ
Der Sänger Sean Levert starb 2008 an den Folgen der Sarkoidose. ⓘ
Joseph Rago, der mit dem Pulitzer-Preis ausgezeichnete Schriftsteller, der für seine Arbeit beim Wall Street Journal bekannt war, starb 2017 an Sarkoidose-Komplikationen. ⓘ
Mehrere historische Persönlichkeiten stehen im Verdacht, an Sarkoidose erkrankt zu sein. In einem Brief an die britische medizinische Fachzeitschrift The Lancet aus dem Jahr 2014 wurde die Vermutung geäußert, dass der französische Revolutionsführer Maximilien Robespierre an Sarkoidose erkrankt gewesen sein könnte, die ihn während seiner Zeit als Anführer der Schreckensherrschaft beeinträchtigte. Die Symptome im Zusammenhang mit Ludwig van Beethovens Tod im Jahr 1827 wurden als möglicherweise mit Sarkoidose vereinbar beschrieben. Der Schriftsteller Robert Louis Stevenson (1850-1894) litt unter chronischem Husten und Brustbeschwerden, und Sarkoidose wurde als Diagnose vorgeschlagen. ⓘ
Schwangerschaft
Sarkoidose steht einer erfolgreichen Schwangerschaft und Entbindung im Allgemeinen nicht im Wege; der Anstieg des Östrogenspiegels während der Schwangerschaft kann sogar eine leicht positive immunmodulatorische Wirkung haben. In den meisten Fällen wird der Krankheitsverlauf durch die Schwangerschaft nicht beeinflusst, in einigen wenigen Fällen kommt es zu einer Besserung, in sehr wenigen Fällen zu einer Verschlechterung der Symptome. Es ist jedoch zu beachten, dass einige der Immunsuppressiva (wie Methotrexat, Cyclophosphamid), die bei kortikosteroidrefraktärer Sarkoidose eingesetzt werden, bekanntermaßen teratogen sind. Für Präeklampsie/Eklampsie, Kaiserschnitt oder Frühgeburt sowie (nicht kardiale) Geburtsfehler bei ersten Einlingsschwangerschaften wurde ein erhöhtes Risiko im Zusammenhang mit Sarkoidose von 30 bis 70 % berichtet. In absoluten Zahlen sind Geburtsfehler und andere Komplikationen wie Tod der Mutter, Herzstillstand, Plazentaablösung oder venöse Thromboembolien bei Sarkoidose-Schwangerschaften extrem selten. ⓘ
Pathogenese
Eine erhöhte inflammatorische Aktivität und eine gesteigerte zelluläre Immunantwort mit Entstehung von nicht einschmelzenden Granulomen bilden die Pathogenese. Diese Granulome zeigen differenzierte Epitheloid- und Riesenzellen. Der beschriebenen Entzündungsreaktion liegt eine Störung der T-Lymphozytenfunktion bei gleichzeitig erhöhter B-Lymphozytenaktivität zugrunde. Dabei kommt es zu einer lokalen immunologischen Überaktivität mit oben beschriebener Granulombildung insbesondere im Lungengewebe und dem lymphatischen System. Besonders betroffen sind Lymphknoten (90 % der Fälle, Lymphknotensarkoidose) sowie die Lunge (90 %, Lungensarkoidose). Aber auch andere Organe wie Leber (60–90 %, Lebersarkoidose), Augen (25 %, Augensarkoidose), Herz (5 %, Herzsarkoidose), Skelett (25–50 %, Skelettsarkoidose), Milz (50–60 %, Milzsarkoidose) oder Haut (25–50 %, Hautsarkoidose) und das Knochenmark (15–40 %) können betroffen sein. Ist das Nervengewebe befallen, so spricht man von einer Neurosarkoidose. Da die Erkrankung familiär gehäuft auftreten kann, wird eine genetische Veranlagung vermutet. Im Februar 2005 wurde eine erste Genveränderung gefunden, die mit einem Ausbrechen der Krankheit korreliert. So reicht die Mutation eines einzigen Basenpaars im Gen BTNL2 auf Chromosom 6 aus, um die Erkrankungswahrscheinlichkeit um 60 % zu erhöhen. Eine Veränderung der Genkopien auf beiden Chromosomen erhöht das Risiko auf das Dreifache. BTNL2 beeinflusst eine Entzündungsreaktion, die bestimmte weiße Blutkörperchen aktiviert. ⓘ
Differentialdiagnose
Die Diagnose erfolgt als Ausschlussdiagnose. Der Verdacht auf Sarkoidose muss vor allem von einer Lungentuberkulose, einer Tumoraussaat der Lunge (Lymphangiosis carcinomatosa) oder einem Lymphom abgegrenzt werden. Auch andere fibrosierende Lungenerkrankungen wie Langerhans-Zell-Histiozytose, exogen-allergische Alveolitis und Pneumokoniosen wie Silikose, Berylliose, Mischstaubsilikose oder Asbestose kommen in Frage. Bei Vorhandensein eines Erythema nodosum muss auch an Borreliose, Yersinien, diverse Bakterien, Mukoviszidose, Morbus Crohn, Lupus erythematodes und weitere Erkrankungen gedacht werden, die mit derselben Symptomatik einhergehen. ⓘ
Mögliche Mitursachen
Die Manifestationsorte Lunge, Haut und Augen lassen ein exogenes Agens erwarten, beispielsweise anorganische Staubbestandteile. So waren Feuerwehrleute vom Ground Zero häufiger von Granulomen betroffen. Auch organische Stoffe, wie beispielsweise bakterielle DNA, wurden in Granulomen gefunden. ⓘ
Gewisse Ähnlichkeiten zwischen Sarkoidose und Tuberkulose führten zum Verdacht, dass auch die Sarkoidose bakterielle Ursachen haben könnte. Versuche, die Sarkoidose mit Tuberkulosemedikamenten zu behandeln, blieben weitgehend erfolglos. In einer Metastudie wurden alle Arbeiten zwischen 1980 und 2006 zusammengefasst, die versuchten, Mykobakterien bei Sarkoidose mithilfe von PCR zu finden. Es ergab sich eine klare Assoziation von manchen Typen der Sarkoidose mit dem Vorhandensein der Erreger. Andere Erreger als Ursache sind nicht ausgeschlossen. ⓘ
In einigen Fällen ist die Auslösung einer Sarkoidose durch Interferon beschrieben, was zu einer relativen Kontraindikation dieses Medikaments bei Sarkoidose geführt hat. ⓘ