Ehlers-Danlos-Syndrom
| Ehlers-Danlos-Syndrome (EDS) ⓘ | |
|---|---|
.png) | |
| Person mit EDS, die eine Hyperelastizität der Haut aufweist | |
| Aussprache |
|
| Fachgebiet | Medizinische Genetik, Rheumatologie |
| Symptome | Übermäßig flexible Gelenke, dehnbare Haut, abnorme Narbenbildung |
| Komplikationen | Aortendissektion, Gelenkverrenkungen, Osteoarthritis |
| Übliches Auftreten | Je nach Typ in der Kindheit oder im Jugendalter. |
| Dauer | Lebenslang |
| Arten | Hypermobile, klassische, vaskuläre, Kyphoskoliose, Arthrochalasie, Dermatosparaxis, Syndrom der brüchigen Hornhaut, andere |
| Ursachen | Genetisch bedingt |
| Risikofaktoren | Familienanamnese |
| Diagnostische Methode | Genetische Tests, körperliche Untersuchung |
| Differentialdiagnose | Marfan-Syndrom, Cutis-Laxa-Syndrom, familiäres Gelenkhypermobilitätssyndrom, Loeys-Dietz-Syndrom, Hypermobilitätsspektrumsstörung |
| Behandlung | Unterstützend |
| Vorhersage | Abhängig von der spezifischen Störung |
| Häufigkeit | 1 von 5.000 |
Bei den Ehlers-Danlos-Syndromen (EDS) handelt es sich um eine Gruppe von dreizehn genetisch bedingten Bindegewebsstörungen, die in der aktuellen Klassifizierung enthalten sind. 2018 wurde ein vierzehnter Typ entdeckt. Zu den Symptomen können lockere Gelenke, Gelenkschmerzen, dehnbare, samtige Haut und abnorme Narbenbildung gehören. Diese können bei der Geburt oder in der frühen Kindheit auftreten. Zu den Komplikationen können Aortendissektion, Gelenkverrenkungen, Skoliose, chronische Schmerzen oder frühe Osteoarthritis gehören. ⓘ
EDS tritt aufgrund von Variationen von mehr als 19 Genen auf, die bei der Geburt vorhanden sind. Das spezifisch betroffene Gen bestimmt die Art des EDS. Einige Fälle entstehen durch eine neue Variation, die während der frühen Entwicklung auftritt, während andere autosomal dominant oder rezessiv vererbt werden. Typischerweise führen diese Variationen zu Defekten in der Struktur oder Verarbeitung des Proteins Kollagen. ⓘ
Die Diagnose basiert häufig auf den Symptomen und wird durch Gentests oder eine Hautbiopsie bestätigt, doch kann es vorkommen, dass die Betroffenen zunächst fälschlicherweise mit Hypochondrie, Depression oder chronischem Müdigkeitssyndrom diagnostiziert werden. ⓘ
Eine Heilung ist derzeit nicht bekannt, und die Behandlung ist eher unterstützend. Physikalische Therapie und Bandagen können helfen, die Muskeln zu stärken und die Gelenke zu stützen. Während einige Formen von EDS zu einer normalen Lebenserwartung führen, ist sie bei den Formen, die die Blutgefäße betreffen, im Allgemeinen geringer. ⓘ
Die hypermobile Form des EDS (hEDS) betrifft weltweit mindestens einen von 5 000 Menschen; andere Formen treten mit geringerer Häufigkeit auf. Die Prognose hängt von der jeweiligen Störung ab. Übermäßige Mobilität wurde erstmals 400 v. Chr. von Hippokrates beschrieben. Die Syndrome sind nach den beiden Ärzten Edvard Ehlers und Henri-Alexandre Danlos benannt, die sie um die Jahrhundertwende beschrieben. ⓘ
| Klassifikation nach ICD-10 ⓘ | |
|---|---|
| Q79.6 | Ehlers-Danlos-Syndrom |
| ICD-10 online (WHO-Version 2019) | |
Das Ehlers-Danlos-Syndrom (EDS) zählt zu den seltenen Krankheiten. Genauer gesagt, ist EDS ein Sammelbegriff für eine heterogene Gruppe von seltenen angeborenen Störungen im Bindegewebe. Die wichtigsten sichtbaren gemeinsamen Erkennungsmerkmale dieser Gruppe sind eine Überdehnbarkeit der Haut und überbewegliche Gelenke. Betroffen sind jedoch auch Gefäße, Muskeln, Bänder, Sehnen und innere Organe. Die Erkrankung kann durch das schwache Bindegewebe tödlich verlaufen, beispielsweise durch spontane Organrupturen, Aortendissektion oder Aneurysmen. ⓘ
Die Erkrankung verläuft in den meisten Fällen fortschreitend. ⓘ
Bisher sind neunzehn Genmutationen bekannt, die EDS auslösen. Die verschiedenen Mutationen führen zu einer Veränderung der Struktur, der Produktion oder der Verarbeitung von Kollagen oder von Proteinen, die mit Kollagen interagieren. Die Häufigkeit des Auftretens in der Bevölkerung wird mit 1:20.000 angenommen, somit handelt es sich bei EDS um eine seltene Erkrankung. Der häufigste Typ ist der hypermobile Typ mit einer Prävelenz von 1:5.000. Die seltensten Typen treten nur wenige Male weltweit auf. Es gibt keine Unterschiede im Auftreten zwischen verschiedenen ethnischen Gruppen oder den Geschlechtern. ⓘ


Arten
Im Jahr 2017 wurden 13 Subtypen von EDS anhand spezifischer Diagnosekriterien klassifiziert. Nach Angaben der Ehlers-Danlos-Gesellschaft können die Syndrome auch nach den Symptomen eingeteilt werden, die durch bestimmte Genmutationen bestimmt werden. Störungen der Gruppe A sind solche, die die primäre Kollagenstruktur und -verarbeitung betreffen. Störungen der Gruppe B betreffen die Kollagenfaltung und -vernetzung. Gruppe C sind Störungen der Struktur und Funktion der Myomatrix. Bei den Störungen der Gruppe D handelt es sich um solche, die die Glykosaminoglykan-Biosynthese betreffen. Die Störungen der Gruppe E sind durch Defekte im Komplementweg gekennzeichnet. Gruppe F sind Störungen intrazellulärer Prozesse, und Gruppe G gilt als ungelöste Form des EDS. ⓘ
Hypermobiles EDS
Hypermobiles EDS (hEDS, früher als Typ 3 kategorisiert) ist hauptsächlich durch Hypermobilität gekennzeichnet, die sowohl große als auch kleine Gelenke betrifft. Es kann zu häufigen Gelenksubluxationen (Teilverrenkungen) und Verrenkungen führen. Im Allgemeinen haben Menschen mit dieser Variante eine weiche, glatte und samtige Haut, die leicht blaue Flecken bekommt, und können unter chronischen Muskel- und/oder Knochenschmerzen leiden. Die Haut ist weniger betroffen als bei anderen Formen. Für diese Variante gibt es keinen Gentest. hEDS ist die häufigste der 19 Arten von Bindegewebserkrankungen. Da es keinen Gentest gibt, müssen die behandelnden Ärzte die Diagnose hEDS auf der Grundlage dessen stellen, was sie bereits über die Krankheit wissen, und anhand der körperlichen Merkmale des Patienten. Neben den allgemeinen Anzeichen können auch fehlerhaftes Bindegewebe im gesamten Körper, Probleme mit dem Bewegungsapparat und eine familiäre Vorgeschichte eine Rolle spielen. Neben diesen allgemeinen Anzeichen und Nebenwirkungen können die Patienten auch Probleme bei der Heilung haben. ⓘ
Frauen, die schwanger sind, sollten vor Dingen wie einem Blasensprung vor der Geburt, einem Blutdruckabfall bei Narkose, einer überstürzten Geburt (sehr schnelle, aktive Wehen), einer Fehlstellung der Blutung und vielem mehr gewarnt werden. Frischgebackene Mütter mit hEDS sollten besonders auf die Pflege ihres Neugeborenen achten. Die Mütter können Schwierigkeiten haben, sich um das Baby zu kümmern, weil sie aufgrund des schwachen Bindegewebes in Armen und Beinen Gefahr laufen, das Baby fallen zu lassen, zu stürzen, postpartale Depressionen zu bekommen (häufiger als in der Allgemeinbevölkerung) und sich vom Geburtsvorgang zu erholen. ⓘ
Genetik des hypermobilen EDS Während es bei 12 der 13 Subtypen von EDS genetische Variationen gibt, die durch Gentests nachgewiesen werden können, ist derzeit keine genetische Ursache für hEDS bekannt. Trotz des unbekannten Gens scheint hEDS einem autosomal-dominanten Vererbungsmuster zu folgen. Das bedeutet, dass eine Person nur eine Mutation in einer Kopie des verantwortlichen (unbekannten) Gens in jeder Zelle haben muss, um betroffen zu sein. Diese Mutation kann entweder von einem Elternteil vererbt werden oder es kann sich um eine de novo (neue) Mutation im Gen handeln. Bei einer Person mit hEDS besteht eine 50%ige Chance, dass sie ihr mutiertes Gen an ihre Kinder weitergibt. ⓘ
In letzter Zeit haben mehrere Labors und Forschungsinitiativen versucht, ein mögliches hEDS-Gen zu entdecken. Im Jahr 2018 begann die Ehlers-Danlos-Gesellschaft mit der Studie Hypermobile Ehlers-Danlos Genetic Evaluation (HEDGE). Im Rahmen der laufenden Studie wurden über 1000 Personen, bei denen nach den Kriterien von 2017 hEDS diagnostiziert wurde, untersucht, um ihr Genom auf eine mögliche gemeinsame Mutation zu untersuchen. Bisher wurden nur bei 200 Personen mit hEDS eine Ganzgenomsequenzierung und bei 500 Personen eine Ganz-Exom-Sequenzierung durchgeführt; mit dieser Studie sollen diese Zahlen deutlich erhöht werden. ⓘ
Das vielversprechendste Ergebnis dieses erweiterten Screenings stammt aus dem Norris-Labor unter der Leitung von Dr. Russell Norris, PhD, in der Abteilung für Regenerative Medizin und Zellbiologie an der Medical University of South Carolina. Mit Hilfe von CRISPR Cas-9 vermitteltem Genome Editing an Mausmodellen der Krankheit hat das Labor kürzlich ein "sehr starkes Kandidatengen" für hEDS identifiziert. Diese Entdeckung und ein besseres Verständnis der kardialen Komplikationen, die mit den meisten EDS-Subtypen einhergehen, haben zur Entwicklung mehrerer medikamentös behandelbarer Signalwege geführt, die an Aorten- und Mitralklappenerkrankungen beteiligt sind. Obwohl dieses Kandidatengen noch nicht öffentlich identifiziert wurde, hat das Norris-Labor mehrere Studien durchgeführt, bei denen kleine Populationen genomisch sequenziert wurden, und hat eine Arbeitsliste möglicher hEDS-Gene erstellt. In einer einzigen Familie mit autosomal dominantem hEDS-Phänotyp wurde eine Mutation in COL3A1 gefunden, die zu einer verminderten Kollagensekretion und einer Übermodifikation von Kollagen führt. In 35 Familien wurden Kopienzahlveränderungen in TPSAB1, das für Alpha-Tryptase kodiert, mit erhöhten Tryptase-Basalwerten im Serum in Verbindung gebracht, die mit autonomen Funktionsstörungen, gastrointestinalen Störungen, allergischen und kutanen Symptomen sowie Bindegewebsanomalien einhergehen, die alle mit dem hEDS-Phänotyp einhergehen. Schließlich wurde Tenascin X, ein Protein der extrazellulären Matrix, das für die Kollagenbildung wichtig ist und vom TNXB-Gen kodiert wird, bei Patienten mit Tenascin-X-Mangel mit hEDS in Verbindung gebracht. ⓘ
Das Norris-Labor versucht auch, dieses schwer fassbare Gen zu finden, indem es Gene untersucht, die an der Bildung der Aorta und der Mitralklappen beteiligt sind, da diese Klappen als Symptom von EDS häufig vorfallen oder missgebildet sind. Da es sich bei hEDS um eine so komplexe, mehrere Organe betreffende Krankheit handelt, hat sich die Konzentration auf ein Merkmal als erfolgreich erwiesen. Ein potenzielles Gen, das auf diesem Weg gefunden wurde, ist DZIP1, das die Herzklappenentwicklung bei Säugetieren über einen Cby1-beta-Catenin-Mechanismus reguliert. Mutationen in diesem Gen wirken sich auf die Beta-Catenin-Kaskade aus, die an der Entwicklung beteiligt ist, und führen zu einer Fehlbildung der extrazellulären Matrix, die einen Verlust von Kollagen zur Folge hat. Ein Mangel an Kollagen steht im Einklang mit hEDS und erklärt den "schlaffen" Charakter der Mitral- und Aortenklappenfehler. Eine zweite genetische Studie, die sich speziell mit dem Mitralklappenprolaps befasst, konzentrierte sich auf den PDGF-Signalweg, der an Wachstumsfaktor-Liganden und Rezeptor-Isoformen beteiligt ist (15). Mutationen in diesem Signalweg wirken sich auf die Fähigkeit zur Lokalisierung von Zilien in verschiedenen Zelltypen, einschließlich Herzzellen, aus. Bei den daraus resultierenden Ziliopathien sind Strukturen wie der kardiale Ausflusstrakt, der Aufbau der Herzröhre und die Herzfusion eingeschränkt und/oder beschädigt. ⓘ
Klassisches EDS
Das klassische EDS (früher als Typ 1 eingestuft) ist gekennzeichnet durch eine extrem elastische Haut, die zerbrechlich ist und leicht Druckstellen bekommt, sowie durch eine Hypermobilität der Gelenke. Molluskoide Pseudotumore (verkalkte Hämatome, die über Druckstellen auftreten) und Sphäroide (fetthaltige Zysten, die über Unterarmen und Schienbeinen auftreten) werden ebenfalls häufig beobachtet. Eine Nebenkomplikation der Hyperelastizität, die in vielen Fällen von EDS auftritt, erschwert das selbstständige Schließen von Wunden. Manchmal ist die motorische Entwicklung verzögert und es kommt zu Hypotonie. Die Variation, die diese Art von EDS verursacht, liegt in den Genen COL5A2, COL5A1 und seltener COL1A1. Es betrifft die Haut stärker als hEDS. Bei klassischem EDS sind oft große Unterschiede in der Darstellung der Symptome von Patient zu Patient zu beobachten. Aufgrund dieser Unterschiede wird EDS häufig unterdiagnostiziert. Ohne Gentests kann medizinisches Fachpersonal auf der Grundlage einer sorgfältigen Untersuchung von Mund, Haut und Knochen sowie durch neurologische Untersuchungen eine vorläufige Diagnose stellen. Die Hyperelastizität der Haut bei EDS-Patienten kann für die Diagnose schwierig sein, da keine gute standardisierte Methode zur Messung und Bewertung der Hautelastizität bekannt ist, aber Hyperelastizität ist dennoch ein guter Indikator, der zusammen mit anderen Symptomen auf EDS hinweisen kann. ⓘ
Eine gute Möglichkeit, den Diagnoseprozess zu beginnen, ist die Untersuchung der Familienanamnese; EDS ist eine autosomal dominante Erkrankung und wird daher häufig von Familienmitgliedern vererbt. Gentests sind nach wie vor der zuverlässigste Weg, um eine EDS-Diagnose zu stellen. Es gibt zwar keine Heilung für EDS Typ 1, aber eine Reihe von Übungen ohne Gewichtsbelastung kann helfen, die Muskelverspannungen zu lösen, wodurch sich einige der EDS-Symptome bessern können. Entzündungshemmende Medikamente und eine Änderung der Lebensweise können bei Gelenkschmerzen helfen. Bei Kindern mit EDS sollte auch der Lebensstil geändert werden, um Wunden an der Haut zu vermeiden. Das Tragen von Schutzkleidung kann dabei helfen. Bei einer Wunde werden oft tiefe Stiche verwendet, die länger als normal an Ort und Stelle verbleiben. ⓘ
Vaskuläre Variante des Ehlers-Danlos-Syndroms
Die vaskuläre Variante des Ehlers-Danlos-Syndroms (früher als Typ 4 eingestuft) zeichnet sich durch eine dünne, durchscheinende Haut aus, die extrem zerbrechlich ist und leicht Druckstellen aufweist. Es ist auch durch brüchige Blutgefäße und Organe gekennzeichnet, die leicht reißen können. Betroffene sind häufig klein und haben dünnes Kopfhaar. Zu den charakteristischen Gesichtszügen gehören große Augen, ein unterdimensioniertes Kinn, eingefallene Wangen, eine dünne Nase und Lippen sowie Ohren ohne Lappen. Eine Hypermobilität der Gelenke ist vorhanden, beschränkt sich aber im Allgemeinen auf die kleinen Gelenke (Finger, Zehen). Weitere häufige Merkmale sind Klumpfuß, Sehnen- und/oder Muskelrisse, Akrogerie (vorzeitige Alterung der Haut an Händen und Füßen), früh einsetzende Krampfadern, Pneumothorax (Zusammenbruch einer Lunge), Zahnfleischrückgang und eine verminderte Menge an Fett unter der Haut. Sie kann durch Variationen im COL3A1-Gen verursacht werden. In seltenen Fällen können auch COL1A1-Variationen die Ursache sein. ⓘ
Kyphoskoliose EDS
Kyphoskoliose EDS (früher als Typ 6 eingestuft) geht mit schwerer Hypotonie bei der Geburt, verzögerter motorischer Entwicklung, progressiver Skoliose (von Geburt an vorhanden) und Sklerafragilität einher. Die Betroffenen leiden auch an leichten Blutergüssen, brüchigen Arterien, die leicht reißen, ungewöhnlich kleinen Hornhäuten und Osteopenie (geringer Knochendichte). Weitere häufige Merkmale sind ein "marfanoider Habitus", der durch lange, schlanke Finger (Arachnodaktylie), ungewöhnlich lange Gliedmaßen und eine eingesunkene Brust (Pectus excavatum) oder einen vorstehenden Brustkorb (Pectus carinatum) gekennzeichnet ist. Sie kann durch Variationen im Gen PLOD1 oder seltener im Gen FKBP14 verursacht werden. ⓘ
Arthrochalasie EDS
Arthrochalasie EDS (früher als Typ 7A und B kategorisiert) ist durch schwere Gelenkhypermobilität und angeborene Hüftluxation gekennzeichnet. Weitere häufige Merkmale sind brüchige, elastische Haut mit leichten Blutergüssen, Hypotonie, Kyphoskoliose (Kyphose und Skoliose) und leichte Osteopenie. In der Regel ist Typ-I-Kollagen betroffen. Sie ist sehr selten, etwa 30 Fälle sind bekannt. Sie ist schwerwiegender als die hypermobile Form. Sie wird durch Variationen in den Genen COL1A1 und COL1A2 verursacht. ⓘ
Dermatosparaxis EDS
Dermatosparaxis EDS (früher als Typ 7C kategorisiert) ist mit extrem brüchiger Haut verbunden, die zu schweren Blutergüssen und Narbenbildung führt; schlaffe, überflüssige Haut, insbesondere im Gesicht; Hypermobilität von leicht bis schwer; und Hernien. Sie wird durch Variationen im ADAMTS2-Gen verursacht. Die Krankheit ist extrem selten, es wurden nur etwa 11 Fälle gemeldet. ⓘ
Glasknochensyndrom
Das Hornhautbrüchigkeitssyndrom ist gekennzeichnet durch eine fortschreitende Ausdünnung der Hornhaut, einen früh einsetzenden progressiven Keratoglobus oder Keratokonus, Kurzsichtigkeit, Hörverlust und blaue Skleren. Klassische Symptome wie hypermobile Gelenke und hyperelastische Haut werden ebenfalls häufig beobachtet. Es gibt zwei Typen. Typ 1 tritt aufgrund von Variationen im ZNF469-Gen auf. Typ 2 ist auf Variationen im PRDM5-Gen zurückzuführen. ⓘ
Klassisches EDS
Das klassisch-ähnliche EDS ist gekennzeichnet durch eine Überdehnbarkeit der Haut mit samtiger Hauttextur und fehlender atrophischer Narbenbildung, eine generalisierte Gelenkhypermobilität mit oder ohne wiederkehrende Luxationen (am häufigsten Schulter und Knöchel) sowie eine leicht zu blauen Flecken neigende Haut oder spontane Ekchymosen (Verfärbungen der Haut durch darunter liegende Blutungen). Sie kann durch Variationen im TNXB-Gen verursacht werden. ⓘ
Spondylodysplastisches EDS
Spondylodysplastisches EDS ist gekennzeichnet durch Kleinwuchs (in der Kindheit fortschreitend), Muskelhypotonie (von schwerer angeborener bis zu leichter späterer Ausprägung) und Verkrümmung der Gliedmaßen. Es kann durch Variationen in beiden Kopien des B4GALT7-Gens verursacht werden. Andere Fälle können durch Variationen im B3GALT6-Gen verursacht werden. Menschen mit Variationen in diesem Gen können Kyphoskoliose, spitz zulaufende Finger, Osteoporose, Aortenaneurysmen und Probleme mit der Lunge haben. Andere Fälle können durch das SLC39A13-Gen verursacht werden. Menschen mit Variationen in diesem Gen haben hervortretende Augen, faltige Handflächen, spitz zulaufende Finger und eine Hypermobilität der distalen Gelenke. ⓘ
Muskulokontraktisches EDS
Das muskulokontrakturelle EDS ist gekennzeichnet durch angeborene multiple Kontrakturen, insbesondere Adduktions-Flexions-Kontrakturen und/oder Talipes equinovarus (Klumpfuß), charakteristische kraniofaziale Merkmale, die bei der Geburt oder im frühen Säuglingsalter deutlich werden, und Hautmerkmale wie Überdehnbarkeit der Haut, Blutergüsse, Hautbrüchigkeit mit atrophischen Narben und verstärkte Faltenbildung an den Handflächen. Sie kann durch Variationen im CHST14-Gen verursacht werden. Einige andere Fälle können durch Variationen im DSE-Gen verursacht werden. ⓘ
Myopathisches EDS
Das myopathische EDS (mEDS) ist durch drei Hauptkriterien gekennzeichnet: angeborene Muskelhypotonie und/oder Muskelatrophie, die sich mit zunehmendem Alter bessert, proximale Gelenkkontrakturen an Knie, Hüfte und Ellbogen sowie Hypermobilität der distalen Gelenke (Knöchel, Handgelenke, Füße und Hände). Außerdem können vier weitere Kriterien zur Diagnose von mEDS beitragen. Diese Störung kann entweder autosomal dominant oder autosomal rezessiv vererbt werden. Molekulare Tests müssen durchgeführt werden, um zu überprüfen, ob Mutationen im COL12A1-Gen vorliegen; falls nicht, sollten andere kollagene Myopathien in Betracht gezogen werden. ⓘ
Parodontales EDS
Das parodontale EDS (pEDS) ist eine autosomal-dominante Erkrankung, die durch vier Hauptkriterien gekennzeichnet ist: schwere und hartnäckige Parodontitis mit frühem Beginn (in der Kindheit oder Jugend), Fehlen von anhaftendem Zahnfleisch, prätibiale Plaques und einen Verwandten ersten Grades in der Familiengeschichte, der die klinischen Kriterien erfüllt. Acht Nebenkriterien können ebenfalls zur Diagnose von pEDS beitragen. Molekulare Tests können Mutationen in C1R- oder C1S-Genen nachweisen, die das C1r-Protein beeinflussen. ⓘ
Kardial-valvuläres EDS
Das kardial-valvuläre EDS (cvEDS) ist durch drei Hauptkriterien gekennzeichnet: schwere, fortschreitende kardial-valvuläre Probleme (die Aorten- und Mitralklappen betreffen), Hautprobleme wie Hyperextensibilität, atrophische Narbenbildung, dünne Haut und leichte Blutergüsse sowie Gelenkhypermobilität (generalisiert oder auf kleine Gelenke beschränkt). Vier kleinere Kriterien können bei der Diagnose von cvEDS hilfreich sein. cvEDS ist eine autosomal rezessive Erkrankung, die durch Variationen in beiden Allelen des Gens COL1A2 vererbt wird. ⓘ
Anzeichen und Symptome
Diese Gruppe von Erkrankungen betrifft das Bindegewebe im gesamten Körper, wobei die Symptome typischerweise an den Gelenken, der Haut und den Blutgefäßen auftreten. Da das Bindegewebe jedoch im ganzen Körper vorkommt, kann EDS zu einer Reihe unerwarteter Auswirkungen mit unterschiedlichem Schweregrad führen, und die Erkrankung ist nicht auf Gelenke, Haut und Blutgefäße beschränkt. Die Auswirkungen können von leicht lockeren Gelenken bis hin zu lebensbedrohlichen kardiovaskulären Komplikationen reichen. Aufgrund der Vielfalt der Subtypen innerhalb der EDS-Familie können die Symptome bei den einzelnen Personen, bei denen EDS diagnostiziert wurde, sehr unterschiedlich sein. ⓘ
Muskuloskelettale Symptome
Zu den muskuloskelettalen Symptomen gehören hyperflexible Gelenke, die instabil sind und zu Verstauchungen, Verrenkungen, Subluxationen und Hyperextensionen neigen. Es kann zu einem frühen Beginn einer fortgeschrittenen Arthrose, einer chronischen degenerativen Gelenkerkrankung, einer Schwanenhalsdeformität der Finger und einer Boutonniere-Deformität der Finger kommen. Es können Sehnen- oder Muskelrisse auftreten. Auch Deformationen der Wirbelsäule wie Skoliose (Wirbelsäulenverkrümmung), Kyphose (Brusthöcker), Tethered Spinal Cord Syndrome, kraniozervikale Instabilität und okzipitoatlantoaxiale Hypermobilität können auftreten. Es können auch Myalgien (Muskelschmerzen) und Arthralgien (Gelenkschmerzen) auftreten, die schwerwiegend und behindernd sein können. Häufig wird das Trendelenburg-Zeichen beobachtet, d. h., wenn man auf einem Bein steht, fällt das Becken auf die andere Seite. Auch die Osgood-Schlatter-Krankheit, eine schmerzhafte Beule am Knie, ist häufig. Bei Säuglingen kann sich das Laufen verzögern (nach dem 18. Lebensmonat), und es kommt zu Bodenbewegungen anstelle des Krabbelns. ⓘ

Person mit EDS mit hypermobilen Fingern, einschließlich der "Schwanenhals"-Fehlbildung an den Ziffern 2-5, und einem hypermobilen Daumen

Person mit EDS, die einen hypermobilen Daumen aufweist

Person mit EDS mit hypermobilen Mittelhandgelenken
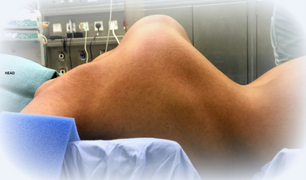
Kyphoskoliose des Rückens bei einer Person mit Kyphoskoliose EDS

Schwere Gelenkhypermobilität bei einem Mädchen mit EDS vom Typ Arthrochalasie ⓘ





Haut
Das schwache Bindegewebe verursacht eine abnorme Haut. Diese kann sich als dehnbar darstellen oder bei anderen Typen einfach samtweich sein. Bei allen Typen kommt es zu einer erhöhten Brüchigkeit, die jedoch je nach zugrunde liegendem Subtyp unterschiedlich stark ausgeprägt ist. Die Haut kann leicht reißen und blaue Flecken bekommen und mit abnormen atrophischen Narben abheilen. Atrophische Narben, die wie Zigarettenpapier aussehen, sind ein Zeichen, das auch bei Menschen beobachtet wird, deren Haut ansonsten normal aussieht. Bei einigen Subtypen, jedoch nicht beim hypermobilen Subtyp, treten überflüssige Hautfalten auf, insbesondere an den Augenlidern. Redundante Hautfalten sind Bereiche mit überschüssiger Haut, die in Falten liegen. ⓘ
Weitere Hautsymptome sind molluskoide Pseudotumore, insbesondere an Druckstellen, Petechien, subkutane Sphäroide, Livedo reticularis und seltener piezoelektrische Papeln. Bei vaskulärem EDS kann die Haut auch dünn und durchscheinend sein. Bei Dermatosparaxis EDS ist die Haut extrem brüchig und schlaff. ⓘ

Atrophische Narbe bei einem EDS-Fall

Transluzente Haut bei vaskulärem EDS

Person mit EDS, die eine Hyperelastizität der Haut aufweist
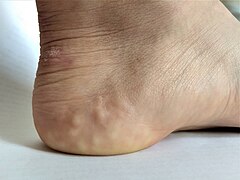
Piezogene Papeln an der Ferse einer Person mit hypermobilem EDS
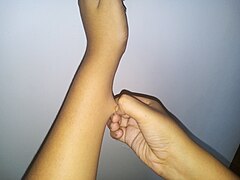
Hyperelastizität der Haut am Handgelenk ⓘ

.png)



Kardiovaskulär
- Thoracic-Outlet-Syndrom
- Arterielle Ruptur
- Herzklappenerkrankungen, wie z. B. ein Mitralklappenprolaps, bergen ein erhöhtes Risiko für eine infektiöse Endokarditis während der Operation. Diese kann einen lebensbedrohlichen Verlauf nehmen. Bei Menschen mit der hypermobilen Form des EDS wurden Herzleitungsanomalien festgestellt.
- Dilatation und/oder Ruptur (Aneurysma) der Aorta ascendens
- Kardiovaskuläre autonome Funktionsstörungen wie das posturale orthostatische Tachykardiesyndrom
- Raynaud-Phänomen
- Krampfadern (Varizen)
- Herzgeräusche
- Anomalien der Erregungsleitung des Herzens ⓘ
Andere Manifestationen
- Hiatalhernie
- Gastroösophagealer Reflux
- Schlechte gastrointestinale Motilität
- Dysautonomie
- Gorlin-Zeichen (Zunge an der Nase berühren)
- Analprolaps
- Plattfüße
- Tracheobronchomalazie / Luftröhrenkollaps
- Kollabierte Lunge (Spontanpneumothorax)
- Nervenstörungen (Karpaltunnelsyndrom, Akroparästhesie, Neuropathie, einschließlich Kleinfaserneuropathie)
- Unempfindlichkeit gegenüber Lokalanästhetika
- Arnold-Chiari-Fehlbildung
- Thrombozytenaggregationsstörung (die Blutplättchen verklumpen nicht richtig)
- Mastzellstörungen (einschließlich Mastzellaktivierungssyndrom und Mastozytose)
- Schwangerschaftskomplikationen: verstärkte Schmerzen, leichte bis mittelschwere peripartale Blutungen, Zervixinsuffizienz, Uterusriss oder vorzeitiger Blasensprung
- Schwerhörigkeit kann bei einigen Typen auftreten
- Auge: Kurzsichtigkeit, Netzhautriss und Netzhautablösung, Keratokonus, blaue Sklera, trockenes Auge, Sjögren-Syndrom, Linsensubluxation, angioide Streifen, Epikanthusfalten, Schielen, Hornhautvernarbung, Hornhautbrüchigkeitssyndrom, Grauer Star, Carotis-Schwellkörperfisteln, Makuladegeneration
- Kraniozervikale Instabilität: verursacht durch Trauma(s) im Kopf- und Halsbereich wie Gehirnerschütterung und Schleudertrauma. Die Bänder im Nacken können nicht richtig heilen, so dass die Nackenstruktur nicht in der Lage ist, den Schädel zu stützen. Dieser kann dann in den Hirnstamm einsinken und den normalen Liquorfluss blockieren, was zu Problemen im Zusammenhang mit dem autonomen Nervensystem führt, das nicht richtig funktioniert.
- Osteoporose und Osteopenie werden mit EDS und symptomatischer Gelenkhypermobilität in Verbindung gebracht
- Es gibt Hinweise darauf, dass EDS häufiger als erwartet mit neurologischen Entwicklungsstörungen wie Aufmerksamkeitsdefizit-Hyperaktivitätsstörung (ADHS) und anderen Lern-, Kommunikations- und motorischen Problemen, einschließlich Autismus und Tourette-Syndrom, in Verbindung gebracht werden kann. ⓘ
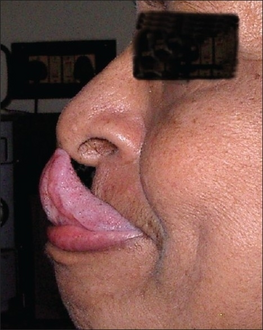
Gorlin-Zeichen in einem Fall von EDS

Keratoglobus bei einem EDS-Fall mit Glaskörpersyndrom ⓘ


Da EDS in der Kindheit häufig nicht oder falsch diagnostiziert wird, wurden einige Fälle von EDS fälschlicherweise als Kindesmisshandlung eingestuft. Die Schmerzen können auch als Verhaltensstörung oder Münchhausen-Syndrom fehldiagnostiziert werden. ⓘ
Die mit EDS verbundenen Schmerzen reichen von leicht bis hin zu schwächend. ⓘ
Ursachen

Jede Art von EDS, mit Ausnahme des hypermobilen Typs (von dem die überwiegende Mehrheit der EDS-Patienten betroffen ist), kann positiv mit einer bestimmten genetischen Variation in Verbindung gebracht werden. ⓘ
Variationen in diesen Genen können EDS verursachen:
- Kollagene Primärstruktur und Kollagenverarbeitung: ADAMTS2, COL1A1, COL1A2, COL3A1, COL5A1, COL5A2
- Kollagenfaltung und Kollagenvernetzung: PLOD1, FKBP14
- Struktur und Funktion der Myomatrix: TNXB, COL12A1
- Glykosaminoglykan-Biosynthese: B4GALT7, B3GALT6, CHST14, DSE
- Komplement-Stoffwechselweg: C1R, C1S
- Intrazelluläre Prozesse: SLC39A13, ZNF469, PRDM5 ⓘ
Variationen in diesen Genen verändern in der Regel die Struktur, die Produktion oder die Verarbeitung von Kollagen oder Proteinen, die mit Kollagen interagieren. Kollagen verleiht dem Bindegewebe Struktur und Festigkeit. Ein Defekt im Kollagen kann das Bindegewebe in der Haut, den Knochen, den Blutgefäßen und den Organen schwächen, was zu den Merkmalen der Erkrankung führt. Die Vererbungsmuster hängen von dem jeweiligen Syndrom ab. ⓘ
Die meisten EDS-Formen werden autosomal-dominant vererbt, was bedeutet, dass nur eine der beiden Kopien des betreffenden Gens verändert sein muss, um eine Störung zu verursachen. Einige wenige werden autosomal-rezessiv vererbt, d. h. beide Kopien des Gens müssen verändert sein, damit eine Person von einer Störung betroffen ist. Es kann sich auch um eine individuelle (de novo oder "sporadische") Veränderung handeln. Sporadische Variationen treten ohne jegliche Vererbung auf. ⓘ
Diagnose
Die Diagnose kann durch Auswertung der Anamnese und der klinischen Beobachtung gestellt werden. Die Beighton-Kriterien werden häufig zur Beurteilung des Ausmaßes der Gelenkhypermobilität herangezogen. DNA- und biochemische Untersuchungen können helfen, die betroffenen Personen zu identifizieren. Zu den diagnostischen Tests gehören Kollagen-Gen-Variantentests, Kollagen-Typisierung durch Hautbiopsie, Echokardiogramm und Lysylhydroxylase- oder Oxidase-Aktivität. Diese Tests sind jedoch nicht in der Lage, alle Fälle zu bestätigen, vor allem wenn es sich um eine nicht kartierte Variante handelt, so dass die klinische Beurteilung wichtig bleibt. Wenn mehrere Personen in einer Familie betroffen sind, kann eine pränatale Diagnose mit Hilfe einer DNA-Informationstechnik, die als Linkage-Studie bekannt ist, durchgeführt werden. Das Wissen über EDS ist bei allen Arten von Ärzten gering. Die Forschung wird fortgesetzt, um genetische Marker für alle Typen zu identifizieren. ⓘ
Differentialdiagnose
Mehrere Erkrankungen haben einige Merkmale mit EDS gemeinsam. Bei der Cutis laxa beispielsweise ist die Haut locker, hängend und faltig. Bei EDS kann die Haut vom Körper weggezogen werden, ist aber elastisch und kehrt in den Normalzustand zurück, wenn sie losgelassen wird. Beim Marfan-Syndrom sind die Gelenke sehr beweglich und es treten ähnliche kardiovaskuläre Komplikationen auf. Menschen mit EDS neigen dazu, ein "marfanoides" Aussehen zu haben (z. B. groß, dünn, lange Arme und Beine, "krakelige" Finger). Die körperliche Erscheinung und die Merkmale verschiedener EDS-Typen weisen jedoch auch Merkmale auf, wie z. B. eine kurze Statur, große Augen und das Erscheinungsbild eines kleinen Mundes und Kinns aufgrund eines kleinen Gaumens. Der Gaumen kann eine hohe Wölbung aufweisen, was zu einem Engstand der Zähne führt. Blutgefäße sind manchmal durch die durchscheinende Haut hindurch leicht zu erkennen, insbesondere auf der Brust. Die genetisch bedingte Bindegewebsstörung, das Loeys-Dietz-Syndrom, weist ebenfalls Symptome auf, die sich mit EDS überschneiden. ⓘ
In der Vergangenheit wurde die Menkes-Krankheit, eine Störung des Kupferstoffwechsels, für eine Form von EDS gehalten. Häufig werden Menschen mit Fibromyalgie, Blutungsstörungen oder anderen Erkrankungen, die EDS-Symptome imitieren können, fehldiagnostiziert. Aufgrund dieser ähnlichen Erkrankungen und der Komplikationen, die bei einem nicht überwachten EDS auftreten können, ist eine korrekte Diagnose wichtig. Pseudoxanthoma elasticum sollte bei der Diagnose in Betracht gezogen werden. ⓘ
Behandlung
Für die Ehlers-Danlos-Syndrome gibt es keine Heilung. Allgemein lässt sich sagen, dass eine auf den Patienten angepasste multimodale Therapie dringend zu empfehlen ist. Diese sollte aus Physiotherapie, Schmerztherapie und psychologischer Unterstützung bestehen. Patienten mit dem vaskulären Typ gelten als am meisten gefährdet und sollten unter ständiger ärztlicher Überwachung stehen. Die medizinische Intervention für alle EDS-Typen ist auf eine symptomatische Therapie begrenzt, die sich in einer Reihe von Empfehlungen auflisten lässt. ⓘ
Eine Überwachung des kardiovaskulären Systems, Physiotherapie, berufliche Rücksichtnahme (sofern das Nachgehen einer beruflichen Tätigkeit möglich ist), orthopädische Hilfsmittel, wie Orthesen, Bandagen, Gehstöcke, Rollatoren oder Rollstühle, können hilfreich sein. Aktivitäten mit Überstreckung bzw. Blockierung der Gelenke sollten vermieden werden. Notwendige chirurgische Eingriffe sollten mit Bedacht durchgeführt werden. Für die ggf. notwendige Anästhesietechnik gibt es verschiedene mögliche Erschwernisse, wie das Nichtansprechen auf Lokalanästhetika, schwierige Atemwegsverhältnisse, Neigung zu Gefäßeinrissen bei Anlage zentralvenöser Katheter und massive Blutungsereignisse insbesondere bei Bagatelloperationen bei EDS-Patienten mit fragilen Gefäßen. Operative Eingriffe sollten nur in Zentren mit ausreichender Expertise in der Behandlung des EDS durchgeführt werden. Bänderraffungen o. Ä. zur Gelenkstabilisierung führen häufig nicht zum gewünschten Erfolg. In der Physiotherapie sollten Haltungstraining und Stabilitätsübungen mit dem Aufbau der dafür verantwortlichen kleinen Muskeln im Vordergrund stehen. Mit Bandagen können die empfindlichsten Stellen vor Verletzungen geschützt werden. Manche Patienten reagieren auf die Gabe von Vitamin C mit verminderter Schwellungsneigung und verbesserter Wundheilung. Versuche mit biotechnischem Hautersatz bei nicht heilenden Wunden waren bei einzelnen Patienten erfolgreich. ⓘ
Kinder sollten mit Informationen über EDS versorgt werden, sodass sie verstehen können, warum Kontaktsportarten oder andere belastende Freizeitbeschäftigungen vermieden werden sollten. Auch ist es wichtig, die Haltungskontrolle frühzeitig zu fördern, um Schäden durch Fehlhaltungen vorzubeugen. Familienmitglieder, Lehrer und Freunde sollten ebenfalls aktiv informiert werden, damit sie das Kind akzeptieren und ggf. fördern können. ⓘ
Emotionale Unterstützung, Verhaltenstherapie und psychologische Unterstützung helfen den Betroffenen aller Subtypen, die Beeinträchtigung zu akzeptieren oder besser damit zurechtzukommen. Patientenorganisationen können dabei behilflich sein (siehe Weblinks). ⓘ
Das Ehlers-Danlos-Syndrom soll ab dem 1. Juli 2021 dem Heilmittelkatalog mit langfristigem Verordnungsbedarf hinzugefügt und vom Bundesministerium für Gesundheit umgesetzt werden. Dies erleichtert Betroffenen langfristige Verschreibungen für Therapien zu bekommen. ⓘ
Auflistung nach Fachgebieten:
| Kardiologie | Ultraschalluntersuchung jährlich, bei unauffälligem Befund alle 3 Jahre; Betablocker bei vergrößerter Aorta und/oder Bluthochdruck; Behandlung von POTS und kardio-vaskulären Problemen klassisch (wie bei Patienten ohne EDS) ⓘ |
| Rheumatologie/Orthopädie | physiotherapeutische Begleitung; lebenslanges Training für Stabilität und Kraft (funktionales Reha-Programm), Übungen mit minimaler Gelenkbeteiligung und wenig Kraft: Balanceübungen, isometrische Übungen, Wassergymnastik, Tai Chi, Pilates, Vermeidung von Kontaktsportarten, repetitiven Bewegungsabläufen und Überdehnung beim Stretching; Gelenkschutz: Vermeidung von Übergewicht, Schuheinlagen; Bandagen, Kinesiotaping für am meisten betroffene Gelenke; Anpassung des Lebens- und Bewegungsstils; ergonomische Arbeits- und Hilfsmittel: unterstützende Matratzen und Kissen, Schreibutensilien mit Griffverstärkung, Haushaltshilfen; Vermeidung von stabilisierenden Operationen; Gelenksersatz, wenn notwendig; periodische Knochendichtemessung |
| Dermatologie/Sportmedizin | Vermeidung von Traumata/Verletzungen, ggf. tägliche Vitamin-C-Gabe; klebende Bandagen können zu Hautrissen führen; Wundverschluss ohne Spannung und in zwei Lagen mit längerer Liegezeit der Fäden, Zugpflaster gegen breite Narben |
| Zahnmedizin | Untersuchungen und Prophylaxe alle 6 Monate, vermindertes Ansprechen bei Lokalanästhesie beachten |
| Gastroenterologie/Innere Medizin | kleine Mahlzeiten; Behandlung von Magen-Darm-Problemen klassisch; besondere Überwachung in der Schwangerschaft |
| Neurologie | Schmerzbehandlung mit steroiden und nichtsteroiden Analgetika, anderen Non-Opioid-Analgetika und Opioid-Analgetika, Antikonvulsiva, Cannabinoiden, Low Dose Naltrexone (LDN) |
| Psychiatrie | psychologische Unterstützung; Schmerztherapie; Vermittlung von Patientenorganisationen |
| Augenheilkunde | regelmäßige Augenuntersuchung (Netzhaut) |
Zur Stabilisierung der Gelenke kann ein Arzt einen Gips verschreiben. Der Arzt kann eine Person an einen Orthopädietechniker für eine orthopädische Behandlung (Schiene) überweisen. Der Arzt kann auch einen Physio- und/oder Ergotherapeuten hinzuziehen, um die Muskeln zu stärken und den Menschen beizubringen, wie sie ihre Gelenke richtig nutzen und schonen können. ⓘ
Die Wassertherapie fördert die Entwicklung der Muskeln und die Koordination. Bei der manuellen Therapie wird das Gelenk im Rahmen des Bewegungsumfangs und/oder durch Manipulationen sanft mobilisiert. Wenn eine konservative Therapie nicht hilft, kann eine chirurgische Gelenkreparatur erforderlich sein. Es können Medikamente zur Schmerzlinderung oder zur Behandlung von Herz-, Verdauungs- oder anderen Erkrankungen verschrieben werden. Um Blutergüsse zu vermindern und die Wundheilung zu verbessern, haben einige Menschen auf Vitamin C angesprochen. Aufgrund der großen Anzahl von Komplikationen, die bei Menschen mit EDS auftreten können, werden von medizinischem Personal oft besondere Vorsichtsmaßnahmen getroffen. Bei vaskulärem EDS werden Anzeichen von Brust- oder Bauchschmerzen als Trauma-Situationen betrachtet. ⓘ
Cannabinoide und medizinisches Marihuana haben eine gewisse Wirksamkeit bei der Schmerzlinderung gezeigt. ⓘ
Schmerzbehandlung
Die erfolgreiche Behandlung chronischer Schmerzen bei EDS erfordert ein multidisziplinäres Team. Die Möglichkeiten zur Schmerzbehandlung können darin bestehen, die in der Normalbevölkerung angewandten Schmerzbehandlungstechniken zu modifizieren. Es gibt zwei Arten von chronischen Schmerzen. Der erste Typ ist der nozizeptive Typ, der durch eine Verletzung des Gewebes verursacht wird. Der zweite Typ ist der neuropathische Schmerz, der durch abnorme Signale des Nervensystems verursacht wird. In den meisten Fällen ist der Schmerz eine ungleiche Mischung aus beiden. Die Physiotherapie (Bewegungsrehabilitation) hat nachweislich eine positive Wirkung. Sie dient in erster Linie der Stabilisierung der Körpermitte und der Gelenke. Dehnübungen müssen auf langsame und sanfte Dehnungen reduziert werden, um das Risiko von Verrenkungen oder Subluxationen zu verringern. Zu den nützlichen Methoden gehören Haltungsumschulung, Muskelentspannung, Gelenkmobilisierung, Rumpfstabilisierung und manuelle Therapie für überlastete Muskeln. Die kognitive Verhaltenstherapie wird bei allen chronischen Schmerzpatienten eingesetzt, insbesondere bei Patienten mit schweren, chronischen, das Leben beherrschenden Schmerzen, die auf eine Behandlung nicht ansprechen. Sie wurde bisher nicht in klinischen Studien auf ihre Wirksamkeit geprüft. Der derzeitige Stand der Schmerzbehandlung bei EDS wird als unzureichend angesehen. ⓘ
Medikamente
Nichtsteroidale Antirheumatika (NSAIDs) können helfen, wenn die Schmerzen durch eine Entzündung verursacht werden. Die langfristige Einnahme von NSAIDs ist jedoch häufig ein Risikofaktor für gastrointestinale, renale und blutbezogene Nebenwirkungen. Sie können die Symptome des Mastzellaktivierungssyndroms verschlimmern, einer Krankheit, die mit EDS in Verbindung gebracht werden kann. Paracetamol kann verwendet werden, um die blutungsbedingten Nebenwirkungen von NSAIDs zu vermeiden. ⓘ
Opioide haben sich in einigen Fällen von EDS als wirksam bei der Behandlung von akuten und chronischen Schmerzen erwiesen. 112 ⓘ
Lidocain kann nach Subluxationen und schmerzhaftem Zahnfleisch topisch aufgetragen werden. Bei Schmerzen des Bewegungsapparats kann es auch in schmerzhafte Bereiche injiziert werden. ⓘ
Ist der Schmerz neuropathischen Ursprungs, können trizyklische Antidepressiva in niedriger Dosierung, Antikonvulsiva und selektive Noradrenalin-Wiederaufnahmehemmer eingesetzt werden. ⓘ
Behandlung von Verrenkungen und Subluxationen
Wenn es zu einer Luxation oder Subluxation kommt, neigen die Muskeln zu Krämpfen und Stress, was die Schmerzen verstärkt und die Wahrscheinlichkeit verringert, dass sich die Luxation/Subluxation auf natürliche Weise zurückbildet. Zu den Methoden zur Unterstützung eines Gelenks nach einem solchen Vorfall gehört die Verwendung einer Schlinge, um das Gelenk zu fixieren und es zu entspannen. Von orthopädischen Gipsverbänden wird abgeraten, da es zu Schmerzen kommen kann, wenn die nicht entspannten Muskeln immer noch versuchen, sich gegen den Verband zu verkrampfen. Andere Lösungen zur Förderung der Entspannung sind Wärme, sanfte Massagen und mentale Ablenkungen. ⓘ
Operation
Die Instabilität von Gelenken, die zu Subluxationen und Gelenkschmerzen führt, erfordert bei Menschen mit EDS häufig einen chirurgischen Eingriff. Instabilitäten können bei fast allen Gelenken auftreten, am häufigsten jedoch in den unteren und oberen Extremitäten, wobei Handgelenk, Finger, Schulter, Knie, Hüfte und Knöchel am häufigsten betroffen sind. ⓘ
Gängige chirurgische Verfahren sind Gelenkdebridement, Sehnenersatz, Kapsulorrhaphie und Arthroplastik. Nach einem chirurgischen Eingriff können sich der Grad der Stabilisierung, die Schmerzreduzierung und die Zufriedenheit der Betroffenen verbessern, aber ein chirurgischer Eingriff ist keine Garantie für ein optimales Ergebnis; Betroffene und Chirurgen berichten, dass sie mit den Ergebnissen unzufrieden sind. Es besteht Einigkeit darüber, dass eine konservative Behandlung wirksamer ist als eine Operation, zumal die Betroffenen aufgrund der Erkrankung ein zusätzliches Risiko für chirurgische Komplikationen haben. Drei grundlegende chirurgische Probleme ergeben sich aus dem EDS: Die Festigkeit des Gewebes ist vermindert, wodurch sich das Gewebe weniger gut für eine Operation eignet; die Brüchigkeit der Blutgefäße kann während der Operation Probleme verursachen; und die Wundheilung ist oft verzögert oder unvollständig. Wenn Sie einen chirurgischen Eingriff in Erwägung ziehen, sollten Sie sich an einen Chirurgen wenden, der über umfassende Kenntnisse und Erfahrungen in der Behandlung von Menschen mit EDS und Problemen der Gelenkhypermobilität verfügt. ⓘ
Lokalanästhetika, arterielle Katheter und zentrale Venenkatheter verursachen bei Menschen mit EDS ein höheres Risiko der Bildung von Blutergüssen. Manche Menschen mit EDS zeigen auch eine Resistenz gegenüber Lokalanästhetika. Eine Resistenz gegen Lidocain und Bupivacain ist nicht ungewöhnlich, und Mepivacain wirkt bei Menschen mit EDS tendenziell besser. Für Menschen mit EDS gibt es spezielle Empfehlungen für die Anästhesie. Detaillierte Empfehlungen für die Anästhesie und die perioperative Versorgung von Menschen mit EDS sollten genutzt werden, um die Sicherheit zu verbessern. ⓘ
Chirurgische Eingriffe bei Menschen mit EDS erfordern eine sorgfältige Behandlung des Gewebes und eine längere Ruhigstellung danach. ⓘ
Vorhersage
Das Ergebnis für Menschen mit EDS hängt von der spezifischen Art von EDS ab, die sie haben. Die Symptome sind unterschiedlich stark ausgeprägt, selbst bei ein und derselben Erkrankung, und die Häufigkeit von Komplikationen ist unterschiedlich. Manche Menschen haben nur geringe Symptome, während andere im täglichen Leben stark eingeschränkt sind. Extreme Gelenkinstabilität, chronische Schmerzen des Bewegungsapparats, degenerative Gelenkerkrankungen, häufige Verletzungen und Wirbelsäulendeformitäten können die Mobilität einschränken. Schwere Wirbelsäulendeformitäten können die Atmung beeinträchtigen. Bei extremer Gelenkinstabilität kann es bei einfachen Tätigkeiten wie dem Umdrehen im Bett oder dem Drehen eines Türknaufs zu Verrenkungen kommen. Sekundäre Erkrankungen wie autonome Funktionsstörungen oder kardiovaskuläre Probleme, die bei jeder Art von Krankheit auftreten, können die Prognose und Lebensqualität beeinträchtigen. Schwere bewegungsbedingte Behinderungen treten bei hEDS häufiger auf als bei klassischem EDS oder vaskulärem EDS. ⓘ
Obwohl alle Arten von EDS potenziell lebensbedrohlich sind, haben die meisten Menschen eine normale Lebenserwartung. Bei Menschen mit einer Fragilität der Blutgefäße besteht jedoch ein hohes Risiko für tödliche Komplikationen, einschließlich einer spontanen Arterienruptur, die die häufigste Ursache für einen plötzlichen Tod ist. Die mittlere Lebenserwartung in der Bevölkerung mit vaskulärem EDS liegt bei 48 Jahren. ⓘ
EDS ist ein lebenslanger Zustand mit meist progredientem Verlauf. Betroffene sehen sich mit sozialen Hindernissen wegen ihrer Krankheit konfrontiert. Einige Patienten berichten von Ängsten vor schwerwiegenden und schmerzhaften Rupturen, vor Verschlimmerung des Zustandes, vor Arbeitslosigkeit wegen ihrer physischen und emotionalen Lasten sowie vor sozialer Ausgrenzung im Allgemeinen. Eine Gentherapie oder andere Ansätze für eine Heilung sind nicht in Sicht, Studien dazu sind bisher nicht bekannt. ⓘ
Komplikationen
Vaskuläres
- Pseudoaneurysma
- Gefäßläsionen (die Art ist umstritten) aufgrund von Rissen in der Auskleidung der Arterien oder der Verschlechterung von angeborenem dünnem und brüchigem Gewebe
- Vergrößerte Arterien ⓘ
Gastrointestinaler Bereich
- Es besteht ein 50%iges Risiko einer Kolonperforation. ⓘ
Geburtshilfe
- Eine Schwangerschaft erhöht die Wahrscheinlichkeit einer Uterusruptur.
- Die mütterliche Sterblichkeit liegt bei etwa 12 %.
- Uterusblutungen können während der postpartalen Erholung auftreten. ⓘ
Entwicklung von Forschungsdatenbanken und Registern
Die Ehlers-Danlos-Gesellschaft und die Bobby Jones Chiari and Syringomyelia Foundation bieten jeweils internationale Patientenregister an, um die Erforschung der Ehlers-Danlos-Syndrome voranzutreiben. Das globale EDS- und HSD-Register und -Repository der Ehlers-Danlos-Gesellschaft ermöglicht die Gensuche nach hypermobilem EDS und erleichtert die Erforschung der Häufigkeit verwandter Symptome und anderer Erkrankungen. Außerdem sollen die Erfahrungen der Menschen, die mit EDS und HSD leben, weltweit erfasst und neue Formen von EDS oder HSD entdeckt werden. Das internationale Patientenregister der Bobby Jones Chiari and Syringomyelia Foundation soll die Wirksamkeit von Gesundheitsprogrammen und Behandlungen für Chiari, Syringomyelie und andere komplizierende Störungen, die häufig bei EDS auftreten, bewerten. ⓘ
Epidemiologie
Schätzungen zufolge treten Ehlers-Danlos-Syndrome weltweit bei etwa einer von 5.000 Geburten auf. Ursprünglich lag die geschätzte Prävalenz zwischen einer von 250.000 und 500.000 Personen, aber diese Schätzungen erwiesen sich bald als zu niedrig, da mehr über die Störungen erforscht wurde und die Mediziner bei der Diagnose immer geschickter wurden. Aufgrund des breiten Spektrums an Schweregraden, mit denen die Störung auftritt, ist EDS möglicherweise weitaus häufiger als die derzeit angenommene Schätzung. ⓘ
Die Prävalenz der Störungen unterscheidet sich dramatisch. Am häufigsten tritt das hypermobile EDS auf, gefolgt vom klassischen EDS. Die anderen sind sehr selten. Zum Beispiel wurden weltweit weniger als 10 Säuglinge und Kinder mit dermatosparaxis EDS beschrieben. ⓘ
Einige Arten von EDS sind bei aschkenasischen Juden häufiger anzutreffen. Die Wahrscheinlichkeit, Träger eines dermatosparaxen EDS zu sein, liegt in der Allgemeinbevölkerung bei eins zu 2.000, während die Prävalenz dieser Variante bei den aschkenasischen Juden bei eins zu 248 liegt. ⓘ
Geschichte
Bis 1997 umfasste das Klassifizierungssystem für EDS 10 spezifische Typen und erkannte an, dass es noch andere, extrem seltene Typen gibt. Zu diesem Zeitpunkt wurde das Klassifizierungssystem überarbeitet und auf sechs Haupttypen mit beschreibenden Bezeichnungen reduziert. Genetiker erkennen an, dass es weitere Typen dieser Erkrankung gibt, die jedoch nur in einzelnen Familien dokumentiert wurden. Mit Ausnahme der Hypermobilität (Typ 3), dem häufigsten Typ aller 10 Typen, sind einige der spezifischen Variationen identifiziert worden, die durch Gentests genau bestimmt werden können, was aufgrund der großen Variationsbreite in Einzelfällen sehr wertvoll ist. Negative Gentestergebnisse schließen die Diagnose jedoch nicht aus, da nicht alle Variationen entdeckt wurden; daher ist die klinische Präsentation sehr wichtig. ⓘ
Formen von EDS in dieser Kategorie können sich durch weiche, leicht dehnbare Haut, verkürzte Knochen, chronischen Durchfall, Gelenkhypermobilität und -verschiebungen, Blasenruptur oder schlechte Wundheilung äußern. Zu den Vererbungsmustern in dieser Gruppe gehören X-chromosomal rezessiv, autosomal dominant und autosomal rezessiv. Beispiele für andere als die oben genannten verwandten Syndrome, die in der medizinischen Fachliteratur beschrieben werden, sind
- 305200: Typ 5
- 130080: Typ 8 - nicht spezifiziertes Gen, Locus 12p13
- 225310: Typ 10 - nicht spezifiziertes Gen, Locus 2q34
- 608763: Beasley-Cohen-Typ
- 130070: progeroide Form - B4GALT7
- 130090: Typ nicht spezifiziert
- 601776: D4ST1-defizientes Ehlers-Danlos-Syndrom (adduziertes Daumen-Klumpfuß-Syndrom) CHST14 ⓘ
Eine EDS-Klassifikation wurde ab den späten 1960er Jahren versucht. 1986 definierte man dann zehn Typen, die 1988 anlässlich einer Konferenz in Berlin veröffentlicht wurden. Mit fortschreitenden Erkenntnissen auf molekularem und biochemischem Gebiet wurde 1997 EDS neu unterteilt. Die Villefranche-Klassifikation diente einer klinisch-vereinfachten Diagnostik des Ehlers-Danlos-Syndroms und zur Abgrenzung von Erkrankungen, die mit dem EDS überlappen (siehe Tabelle). Es ließ sich nun die Krankheit durch sechs Typen mit ihren Haupt- und Nebenkriterien beschreiben. Darüber wurden weitere andere, exotische Formen von EDS in Einzelfällen identifiziert. Seit 2017 wird die Villefranche-Klassifikation durch die neuen Diagnosekriterien abgelöst. Die Kriterien wurden vollständig überarbeitet und aktualisiert und unterscheiden nun in 13 Typen. Folgende Beschwerdebilder werden nach den neuen Diagnosekriterien nicht mehr dem EDS Spektrum zugeordnet: Okzipitalhorn-Syndrom, Fibronectin-Mangel (EDS Typ X), familiäres Hypermobilitätssyndrom (EDS Typ XI), X-linked EDS (EDS Typ V) und Filamin A EDS. ⓘ
Es besteht eine große Variabilität und Überlappung in den Symptomen zwischen den einzelnen Typen. Eine eindeutige Klassifizierung ist somit nur anhand klinischer Diagnostik und genetischer Tests bzw. Hautbiopsien möglich. ⓘ
Die auslösenden Mutationen wurden identifiziert. Einzig beim hypermobilen Typ ist das auslösende Gen noch nicht bekannt. ⓘ
Das Syndrom ist eine der ältesten bekannten Ursachen von Ergüssen und Blutungen, die schon von Hippokrates 400 v. Chr. erkannt wurden. Eine erste Fallanalyse mit abnormer Hautelastizität eines spanischen Mannes ist durch den niederländischen Chirurgen Job Janzoon von Meerkerin aus dem Jahre 1668 bekannt. Die erste umfassende Beschreibung des Syndroms mit seinen vielen Facetten (Haut, Gelenke, Narben usw.) entstand 1891 durch den russischen Dermatologen Tschernogubow. Das isolierte Zarenreich verhinderte jedoch das allgemeine Bekanntwerden der Studie, sodass Edvard Ehlers mit der Beschreibung der wesentlichen Zusammenhänge 1901 als Cutis laxa und Henri-Alexandre Danlos 1908 mit dem Vorschlag, die Überdehnbarkeit und Zerreißbarkeit der Haut als Kardinalsymptome zu benennen, die Namensgeber des Syndroms wurden. ⓘ
Andere vorwiegend veraltete Bezeichnungen des EDS waren:
- Ehlers-Danlos-Meekeren-Syndrom, Meekeren-Ehlers-Danlos-Syndrom, Van-Meekeren-Syndrom
- Danlos-Syndrom
- Fibrodysplasia elastica generalisata, Cutis hyperelastica
- Dermatolyse
- Gummihaut, Indian rubber skin
- Tschernogubow-Syndrom
- Sack-Barabas-Syndrom ⓘ
Gesellschaft und Kultur
EDS könnte zu den Fähigkeiten des Geigenvirtuosen Niccolò Paganini beigetragen haben, da er in der Lage war, breitere Fingersätze zu spielen als ein typischer Geiger. ⓘ
Viele Varietékünstler haben EDS. Mehrere von ihnen wurden als "Elastic Skin Man", "India Rubber Man" und "Frog Boy" bezeichnet. Zu ihnen gehörten (zu ihrer Zeit) so bekannte Persönlichkeiten wie Felix Wehrle, James Morris und Avery Childs. Zwei Künstler mit EDS halten derzeit Weltrekorde. Der Schlangenmensch Daniel Browning Smith hat hypermobiles EDS und hält seit 2018 den aktuellen Guinness-Weltrekord für den beweglichsten Mann, während Gary "Stretch" Turner, Sideshow-Darsteller im Zirkus des Schreckens, seit 1999 den aktuellen Guinness-Weltrekord für die elastischste Haut hält, da er die Haut seines Bauches um 6,25 Zoll dehnen kann. ⓘ
Bemerkenswerte Fälle


- Die Schauspielerin Cherylee Houston hat hypermobiles EDS. Sie ist auf einen Rollstuhl angewiesen und war die erste behinderte Vollzeitdarstellerin in der Coronation Street
- Dragqueen und Gewinnerin der 11. Staffel von RuPaul's Drag Race Yvie Oddly
- Eric der Schauspieler, ein regelmäßiger Gast in der Howard Stern Show
- Schauspielerin und Aktivistin Jameela Jamil
- Schriftstellerin und Schauspielerin Lena Dunham
- Australische Sängerin Sia
- YouTuberin und Behindertenrechtsaktivistin Annie Elainey
- Miss Amerika 2020 Camille Schrier
- Gehörlose Sängerin/Songschreiberin Mandy Harvey
- Pornodarstellerin und Autorin Lorelei Lee
- Reality-Show-Teilnehmer Trevor Jones, der an der vaskulären Form der Krankheit starb ⓘ
- Die scheinbar alkoholkranke Teenagerin Emma in Folge 4 der 13. Staffel (Langsamer Fall) der Serie Grey’s Anatomy hat EDS.
- In Folge 18 der Staffel 7 der Serie Dr. House hat Nina ein Messie-Syndrom infolge der Fehlgeburt durch EDS.
- In Folge 3 der 3. Staffel von Atlanta Medical hat der verletzte Schwerkriminelle ständige Schmerzen durch EDS.
- Yvie Oddly, Gewinnerin der 11. Staffel der US-amerikanischen Reality-TV-Serie RuPaul’s Drag Race, leidet an EDS.
- Die britische Miss Universe-Anwärterin Saarah Ahmed starb an EDS. ⓘ
Andere Arten
Ehlers-Danlos-ähnliche Syndrome sind bei Himalaya-Katzen, einigen Kurzhaar-Hauskatzen und bestimmten Rinderrassen nachweislich vererbbar. Bei Haushunden tritt es sporadisch auf. Behandlung und Prognose sind ähnlich. Tiere mit dieser Krankheit sollten nicht gezüchtet werden, da die Krankheit vererbt werden kann. ⓘ
- Tierisches EDS

EDS bei einem Hund

Derselbe Hund mit EDS
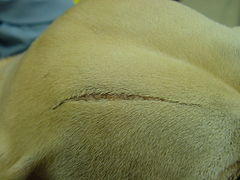
EDS bei demselben Hund mit einer atrophischen Narbe ⓘ


Die degenerative Ligamentum suspensum desmitis ist eine ähnliche Erkrankung, die bei vielen Pferderassen auftritt. Ursprünglich wurde sie beim peruanischen Paso festgestellt und als Folge von Überanstrengung und höherem Alter angesehen, aber sie wird in allen Altersgruppen und auf allen Aktivitätsebenen festgestellt. Sie wurde auch bei neugeborenen Fohlen festgestellt. ⓘ

Ein Ehlers-Danlos-Syndrom tritt auch bei Hunden und Katzen auf. Betroffene Tiere zeigen eine stark erhöhte Verletzbarkeit der Haut, wobei die Hautwunden schnell vernarben. Die Dehnbarkeit der Haut ist stark erhöht. Die Erkrankung wird rezessiv vererbt. Differentialdiagnostisch ist vor allem eine Verdünnung der Haut infolge hoher Kortisolspiegel (Cushing-Syndrom) oder Langzeittherapie mit Glucocorticoiden abzuklären. Die Diagnose wird elektronenmikroskopisch gesichert. Eine Behandlung ist nicht möglich. ⓘ
Diagnostik und Symptomatik

Die EDS-Diagnostik beginnt zunächst mit einer eingehenden ärztlichen Untersuchung. Hierbei sollte zunächst mit Hilfe des Beighton Scores die Gelenküberbeweglichkeit getestet werden. Die Haut wird in Hinblick auf Gefühl (wie teigig oder samtig) und Überdehnbarkeit untersucht. Auch eventuelle Narben sollten begutachtet werden. Außerdem sollten Patienten umfangreich zur Symptomatik befragt und die Krankenakte beachtet werden. Die erhobenen Ergebnisse werden dann mit den klinischen Kriterien der Ehlers-Danlos-Syndrome abgeglichen. Eine finale Diagnose wird durch eine Genuntersuchung gestellt. Die Diagnose hypermobiles Ehlers-Danlos-Syndrom (hEDS) wird klinisch gestellt. Die Diagnosekriterien unterteilen in drei Kriterienbereiche, die alle erfüllt werden müssen. Kriterium 1 ist ein positiver Befund hinsichtlich des Beighton Scores (mindestens 5/9 bei Erwachsenen). Kriterium 2 ist unterteilt in die Bereiche A, B und C. Um Kriterium 2 zu erfüllen, müssen zwei der drei Bereiche zutreffen. A umfasst das Vorliegen von mind. 5/12 systematischen Manifestationen, B umfasst ein familiäres Auftreten von hEDS und C umfasst Schmerzen und Instabilitäten. Kriterium 3 sieht einen Ausschluss anderer Erkrankungen vor. ⓘ
Differentialdiagnostisch sollten u. a. folgende Erkrankungen abgeklärt werden:
- Marfan-Syndrom, Loeys-Dietz-Syndrom Q87.4
- Osteogenesis imperfecta Q87.0
- Stickler-Syndrom Q87.8
- Achondroplasie (Chondrodysplasie) Q77.4
- Rheumatoide Arthritis M06
- Fibromyalgie M79.7
- Multiple Sklerose G35
- Hypermobilitätssyndrom M35.7
- Wachstumsschmerzen R29.8
- Akrogerie E34.8
Die Diagnostizierung von seltenen Krankheiten im Allgemeinen und EDS im Besonderen wirft in der Praxis Probleme auf. Nach einer Studie im Jahr 2005 erhielten z. B. ca. 25 % der Betroffenen ihre EDS-Diagnose erst 28 Jahre nach dem Auftreten der ersten Symptome. ⓘ
Für eine EDS-Diagnose ist die Erstformulierung eines klinischen Verdachtes entscheidend. Ausschlaggebend ist die Präsenz von Major- und Minor-Kriterien, die eine hohe Sensitivität für die Krankheit darstellen. Eine positive Familienhistorie kann den Verdacht auf ein EDS erhärten. In der Praxis gestaltet sich die Diagnosestellung allerdings oft anders. Meist sind es verschiedene Symptome, die in Kombinationen, Schwere und Häufigkeiten des Auftretens ungewöhnlich sind und somit die Suche nach einer erklärenden Ursache initiieren. Dabei stoßen Erkrankte häufig auf Hürden im Gesundheitssystem. Die Unkenntnis über EDS von weiten Teilen der Ärzteschaft lässt die Betroffenen oft eine jahrelange Odyssee bis zur Diagnosestellung erleben. ⓘ
Symptomatiken nach Bereichen (unvollständig): ⓘ
| Kardiologie | Aortenaneurysma; Gefäßerweiterungen; vergrößertes Herzkranzgefäß; Mitralklappenprolaps; Chronische Cerebro-Spinale Venöse Insuffizienz; Posturales orthostatisches Tachykardiesyndrom POTS ⓘ |
| Rheumatologie/Orthopädie | Hypermobilität (Beighton Score ≥ 5); Senkfuß, Knickfuß, Spreizfuß; spontane Sub-/ Luxationen; Chronische Gelenk- und Muskelschmerzen; Skoliose/Kyphose; Beinlängenunterschied; Hüftdysplasie; Hernie; Kielbrust (Pectus carinatum), frühzeitige Arthrose; Degeneration der Bandscheiben; Zysten an Gelenken, Arachnodaktylie, Klinodaktylie des kleinen Fingers. |
| Dermatologie/Sportmedizin | weiche, samtige Haut; dünne, durchscheinende Haut; leichte Verletzbarkeit; breite Narben; kleine Griessknoten; verzögerte Wundheilung; Hämatomneigung; häufige Verletzungen; schnelle Ermüdung; verzögerte motorische Entwicklung; Muskelhypotonie |
| Zahnmedizin | hoher Gaumen; Zahnfehlstellungen; Zahnfleischrückgang; Kiefergelenkschmerzen, kraniomandibuläre Dysfunktion; hypermobile Zunge; Blutungen im Mund-/ Rachenraum |
| Gastroenterologie/Innere Medizin | Eingeweidebrüche (Hernie); Gastroparese; Übelkeit; Abdominalschmerz; Verdauungsstörungen durch Kontraktionsstörungen; Sodbrennen, Reizdarmsyndrom, Dickdarmerweiterungen, Hämorrhoiden, Gebärmuttersenkung, Mastzellaktivitätsstörung; chronische Erschöpfung |
| Neurologie | Chiari-Malformation; Tethered cord; Dysautonomie; Neuralgie; Migräne; zervikale Spondylolisthesis; HWS-Erkrankung; Kopfschmerzen; generelle Koordinationsstörungen; schlechtes Balancegefühl; spinale Stenose; verminderte Wirksamkeit von Lokalanästhetikum; Schwäche; Müdigkeit; Schlafstörung |
| Psychiatrie | Depression; Angstzustand; Gesellschaftsrückzug durch Unverstandensein und Schmerzen; |
| Augenheilkunde | Lichtempfindlichkeit; Schielen; Kurzsichtigkeit; trockene Augen; Netzhautablösung; Linsenverschiebung; Keratokonus; Makuladegeneration; Grüner Star; Grauer Star |