Glioblastom
| Glioblastom ⓘ | |
|---|---|
| Andere Bezeichnungen | Glioblastoma multiforme, Astrozytom Grad IV |
 | |
| Koronales MRT mit Kontrastmittel eines Glioblastoms bei einem 15-jährigen Jungen | |
| Fachgebiet | Neuroonkologie, Neurochirurgie |
| Symptome | Anfänglich unspezifisch, Kopfschmerzen, Persönlichkeitsveränderungen, Übelkeit, schlaganfallähnliche Symptome |
| Gewöhnliches Auftreten | ~ ca. 64 Jahre alt |
| Ursachen | Meist unklar |
| Risikofaktoren | Genetische Störungen (Neurofibromatose, Li-Fraumeni-Syndrom), vorherige Strahlentherapie |
| Diagnostische Methode | CT-Scan, MRI-Scan, Gewebebiopsie |
| Vorbeugung | Unbekannt |
| Behandlung | Operation, Chemotherapie, Bestrahlung |
| Medikation | Temozolomid, Steroide |
| Prognose | Lebenserwartung ~ 12 Monate mit Behandlung (5-Jahres-Überleben <10%) |
| Häufigkeit | 3 pro 100.000 pro Jahr |
Das Glioblastom, früher bekannt als Glioblastoma multiforme (GBM), ist eine der aggressivsten Krebsarten, die im Gehirn entstehen. Anfänglich sind die Anzeichen und Symptome des Glioblastoms unspezifisch. Sie können Kopfschmerzen, Persönlichkeitsveränderungen, Übelkeit und ähnliche Symptome wie bei einem Schlaganfall umfassen. Die Symptome verschlimmern sich oft rasch und können bis zur Bewusstlosigkeit gehen. ⓘ
Die Ursache der meisten Glioblastome ist nicht bekannt. Zu den seltenen Risikofaktoren gehören genetische Störungen wie Neurofibromatose und Li-Fraumeni-Syndrom sowie eine vorangegangene Strahlentherapie. Glioblastome machen 15 % aller Hirntumoren aus. Sie können entweder von normalen Gehirnzellen ausgehen oder sich aus einem bereits bestehenden niedriggradigen Astrozytom entwickeln. Die Diagnose wird in der Regel durch eine Kombination aus CT-Scan, MRT-Scan und Gewebebiopsie gestellt. ⓘ
Es gibt keine bekannte Methode zur Vorbeugung des Krebses. Die Behandlung umfasst in der Regel einen chirurgischen Eingriff, nach dem eine Chemo- und Strahlentherapie durchgeführt wird. Das Medikament Temozolomid wird häufig als Teil der Chemotherapie eingesetzt. Hochdosierte Steroide können zur Verringerung der Schwellung und zur Linderung der Symptome eingesetzt werden. Die chirurgische Entfernung (Dekompression) des Tumors führt zu einer Verlängerung der Überlebenszeit, allerdings nur um einige Monate. ⓘ
Trotz maximaler Behandlung tritt der Krebs fast immer wieder auf. Die typische Überlebensdauer nach der Diagnose beträgt 10-13 Monate, wobei weniger als 5-10 % der Betroffenen länger als fünf Jahre überleben. Ohne Behandlung beträgt die Überlebenszeit in der Regel drei Monate. Es ist der häufigste Krebs, der im Gehirn entsteht, und nach dem Meningeom der zweithäufigste Hirntumor. Etwa 3 von 100 000 Menschen erkranken pro Jahr daran. Das Durchschnittsalter bei der Diagnose liegt bei 64 Jahren, und die Krankheit tritt häufiger bei Männern als bei Frauen auf. ⓘ
| Klassifikation nach ICD-10 ⓘ | |
|---|---|
| C71 | Bösartige Neubildung des Gehirns |
| C71.0 | Zerebrum, ausgenommen Hirnlappen und Ventrikel |
| C71.1 | Frontallappen |
| C71.2 | Temporallappen |
| C71.3 | Parietallappen |
| C71.4 | Okzipitallappen |
| C71.5 | Hirnventrikel |
| C71.6 | Zerebellum |
| C71.7 | Hirnstamm |
| C71.8 | Gehirn, mehrere Teilbereiche überlappend |
| C71.9 | Gehirn, nicht näher bezeichnet |
| ICD-10 online (WHO-Version 2019) | |
Anzeichen und Symptome
Zu den häufigsten Symptomen gehören Krampfanfälle, Kopfschmerzen, Übelkeit und Erbrechen, Gedächtnisverlust, Veränderungen der Persönlichkeit, der Stimmung oder der Konzentration sowie lokal begrenzte neurologische Probleme. Die Art der Symptome hängt eher von der Lage des Tumors als von seinen pathologischen Eigenschaften ab. Der Tumor kann schnell erste Symptome hervorrufen, bleibt aber gelegentlich symptomlos, bis er eine enorme Größe erreicht. ⓘ
Risikofaktoren
Die Ursache der meisten Fälle ist unklar. Etwa 5 % entwickeln sich aus einer anderen Art von Hirntumor, einem so genannten niedriggradigen Astrozytom. ⓘ
Genetik
Glioblastome sind bei Erwachsenen die häufigsten bösartigen hirneigenen Tumoren. Unter den aus dem Hirngewebe entstehenden (neuroepithelialen) Tumoren machen sie etwa die Hälfte aller Fälle aus. Der Tumor tritt am häufigsten bei älteren Erwachsenen zwischen dem 60. und 70. Lebensjahr auf; das durchschnittliche Alter bei Diagnosestellung beträgt 64 Jahre. Männer sind deutlich öfter betroffen als Frauen (Verhältnis 1,7:1). Daten des amerikanischen Hirntumorregisters zeigen, dass Glioblastome bei Weißen mindestens doppelt so häufig sind wie in der schwarzen Bevölkerung. Im Vergleich zu Erwachsenen sind Glioblastome bei Kindern sehr selten. Die Inzidenz wurde in Europa und Nordamerika mit 2,9 bis 3,5 Neuerkrankungen pro Jahr auf 100.000 Einwohner ermittelt und ist in Entwicklungsländern geringer. Als einziger gesicherter ursächlicher (ätiologischer) Umweltfaktor gilt derzeit eine Exposition durch ionisierende Strahlung. ⓘ
Bei der Mehrzahl der Glioblastome handelt es sich um sporadisch auftretende Fälle ohne Hinweis auf eine Erblichkeit. Bei bestimmten seltenen erblichen Erkrankungen, unter anderem bei dem Li-Fraumeni-Syndrom oder dem Turcot-Syndrom, können Glioblastome jedoch in Familien gehäuft auftreten. ⓘ
Umwelt
Weitere Assoziationen bestehen mit Rauchen, Pestiziden und der Arbeit in der Erdölraffinerie oder der Gummiherstellung. ⓘ
Das Glioblastom wurde mit den Viren SV40, HHV-6 und Cytomegalovirus in Verbindung gebracht. ⓘ
Andere
Es wurde untersucht, ob der Verzehr von gepökeltem Fleisch einen Risikofaktor darstellt. Bis zum Jahr 2013 wurde kein Risiko bestätigt. Auch die Exposition gegenüber Strahlung bei medizinischen Bildgebungsverfahren, Formaldehyd und elektromagnetischen Feldern in Wohnräumen, z. B. durch Mobiltelefone und elektrische Leitungen in Häusern, wurden als Risikofaktoren untersucht. Bis zum Jahr 2015 konnte nicht nachgewiesen werden, dass sie GBM verursachen. ⓘ
Pathogenese
Der zelluläre Ursprung des Glioblastoms ist unbekannt. Aufgrund der Ähnlichkeiten in der Immunfärbung von Gliazellen und Glioblastomen wurde lange Zeit angenommen, dass Gliome wie das Glioblastom von Zellen des Glia-Typs abstammen. Neuere Studien legen nahe, dass Astrozyten, Oligodendrozyten-Vorläuferzellen und neurale Stammzellen als Ursprungszellen in Frage kommen. ⓘ
Glioblastome zeichnen sich durch das Vorhandensein kleiner Bereiche mit nekrotisierendem Gewebe aus, die von anaplastischen Zellen umgeben sind. Dieses Merkmal sowie das Vorhandensein von hyperplastischen Blutgefäßen unterscheidet den Tumor von Astrozytomen des Grades 3, die diese Merkmale nicht aufweisen. ⓘ
GBMs bilden sich in der Regel in der weißen Hirnsubstanz, wachsen schnell und können sehr groß werden, bevor sie Symptome verursachen. Weniger als 10 % bilden sich langsamer nach der Degeneration eines niedriggradigen Astrozytoms oder anaplastischen Astrozytoms. Diese werden als sekundäre GBMs bezeichnet und treten häufiger bei jüngeren Patienten auf (Durchschnittsalter 45 versus 62 Jahre). Der Tumor kann in die Hirnhäute oder die Ventrikelwand eindringen, was zu einem hohen Proteingehalt im Liquor (> 100 mg/dl) und gelegentlich zu einer Pleozytose von 10 bis 100 Zellen, meist Lymphozyten, führt. Im Liquor befindliche bösartige Zellen können sich (selten) auf das Rückenmark ausbreiten oder eine meningeale Gliomatose verursachen. Eine Metastasierung des GBM über das zentrale Nervensystem hinaus ist jedoch äußerst selten. Etwa 50 % der GBMs befallen mehr als einen Lappen einer Hemisphäre oder sind bilateral. Tumore dieses Typs entstehen in der Regel im Großhirn und können die klassische Infiltration über den Corpus Callosum aufweisen, was zu einem (bilateralen) Schmetterlingsgliom führt. ⓘ
Klassifizierung von Glioblastomen
Die Klassifizierung von Hirntumoren basiert traditionell auf der Histopathologie auf makroskopischer Ebene, gemessen in Hämatoxylin-Eosin-Schnitten. Die Weltgesundheitsorganisation (WHO) veröffentlichte 1979 die erste Standardklassifikation und hat diese seitdem fortgeführt. Die WHO-Klassifikation von Tumoren des Zentralnervensystems aus dem Jahr 2007 war die letzte Klassifikation, die hauptsächlich auf mikroskopischen Merkmalen beruhte. Die neue WHO-Klassifikation der Tumoren des zentralen Nervensystems von 2016 war ein Paradigmenwechsel: Einige der Tumoren wurden auch durch ihre genetische Zusammensetzung sowie ihre Zellmorphologie definiert. ⓘ
Die Einteilung der Gliome änderte sich grundlegend, und das Glioblastom wurde nun hauptsächlich nach dem Status der Isocitrat-Dehydrogenase (IDH)-Mutation klassifiziert: IDH-Wildtyp oder IDH-mutant. ⓘ
| IDH-Wildtyp-Glioblastom | IDH-mutiertes Glioblastom ⓘ | |
|---|---|---|
| Synonym | Primäres Glioblastom | Sekundäres Glioblastom |
| Vorläufer-Läsion | De novo identifiziert | Diffuses Astrozytom
Anaplastisches Astrozytom |
| Anteil der Glioblastome | ~90% | ~10% |
| Medianes Alter bei der Diagnose | ~62 Jahre | ~44 Jahre |
| Verhältnis Männer:Frauen | 1.42:1 | 1.05:1 |
| Mittlere Dauer der klinischen Vorgeschichte bei der Diagnose | 4 Monate | 15 Monate |
| Medianes Gesamtüberleben | ||
| Chirurgie + Strahlentherapie | 9,9 Monate | 24 Monate |
| Chirurgie + Strahlentherapie + Chemotherapie | 15 Monate | 31 Monate |
| Lage | Supratentoriell | Bevorzugt frontal |
| Nekrose | Ausgedehnt | Begrenzt |
| TERT-Promotor-Mutationen | 72% | 26% |
| TP53-Mutationen | 27% | 81% |
| ATRX-Mutationen | Außergewöhnlich | 71% |
| EGFR-Amplifikation | 35% | Außergewöhnlich |
| PTEN-Mutationen | 24% | Außergewöhnlich |
Molekulare Veränderungen
Auf der Grundlage der Genexpression wurden vier Subtypen des Glioblastoms identifiziert:
- Klassisch: Etwa 97 % der Tumoren dieses Subtyps tragen zusätzliche Kopien des Gens für den epidermalen Wachstumsfaktor-Rezeptor (EGFR), und die meisten haben eine höhere als die normale Expression von EGFR, während das Gen TP53 (p53), das bei Glioblastomen häufig mutiert ist, bei diesem Subtyp selten mutiert ist. Der Verlust der Heterozygotie auf Chromosom 10 ist neben der Amplifikation von Chromosom 7 ebenfalls häufig beim klassischen Subtyp zu beobachten.
- Der proneurale Subtyp weist häufig hohe Raten von Veränderungen in TP53 (p53), in PDGFRA, dem Gen für den Rezeptor des Plättchenwachstumsfaktors vom a-Typ, und in IDH1, dem Gen für Isocitratdehydrogenase-1, auf.
- Der mesenchymale Subtyp ist durch eine hohe Rate an Mutationen oder anderen Veränderungen in NF1, dem Gen, das für Neurofibromin 1 kodiert, sowie durch weniger Veränderungen im EGFR-Gen und eine geringere Expression von EGFR als bei anderen Typen gekennzeichnet.
- Der neuronale Subtyp zeichnet sich durch die Expression von Neuronenmarkern wie NEFL, GABRA1, SYT1 und SLC12A5 aus, während sie sich bei der pathologischen Untersuchung oft als normale Zellen darstellen. ⓘ
Beim Glioblastom wurden zahlreiche weitere genetische Veränderungen beschrieben, von denen die meisten in zwei Signalwegen, dem RB- und dem PI3K/AKT-Signalweg, zusammengefasst sind. Glioblastome weisen Veränderungen in 68-78 % bzw. 88 % dieser Signalwege auf. ⓘ
Eine weitere wichtige Veränderung ist die Methylierung von MGMT, einem "Selbstmord"-DNA-Reparaturenzym. Die Methylierung beeinträchtigt die DNA-Transkription und die Expression des MGMT-Gens. Da das MGMT-Enzym aufgrund seines Selbstmord-Reparaturmechanismus nur eine DNA-Alkylierung reparieren kann, ist die Reservekapazität gering, und die Methylierung des MGMT-Genpromotors beeinträchtigt die DNA-Reparaturkapazität erheblich. Die MGMT-Methylierung steht in Zusammenhang mit einem verbesserten Ansprechen auf die Behandlung mit DNA-schädigenden Chemotherapeutika, wie z. B. Temozolomid. ⓘ
Krebsstammzellen
In Glioblastomen wurden Glioblastomzellen mit ähnlichen Eigenschaften wie Vorläuferzellen (Glioblastomkrebs-Stammzellen) gefunden. Ihr Vorhandensein in Verbindung mit der diffusen Natur des Glioblastoms führt zu der Schwierigkeit, sie durch eine Operation vollständig zu entfernen, und wird daher als mögliche Ursache für die Resistenz gegenüber herkömmlichen Behandlungen und die hohe Rückfallquote angesehen. Glioblastom-Krebsstammzellen weisen eine gewisse Ähnlichkeit mit neuralen Vorläuferzellen auf, da beide den Oberflächenrezeptor CD133 exprimieren. CD44 kann in einer Untergruppe von Glioblastom-Tumorzellen auch als Krebsstammzellmarker verwendet werden. Glioblastom-Krebsstammzellen scheinen eine erhöhte Resistenz gegenüber Strahlen- und Chemotherapie aufzuweisen, die zumindest teilweise durch eine Hochregulierung der DNA-Schadensreaktion vermittelt wird. ⓘ
Stoffwechsel
Das IDH1-Gen kodiert für das Enzym Isocitrat-Dehydrogenase 1 und ist beim Glioblastom selten mutiert (primäres GBM: 5%, sekundäres GBM >80%). Durch die Produktion sehr hoher Konzentrationen des "Onkometaboliten" D-2-Hydroxyglutarat und die Dysregulation der Funktion des IDH1-Wildtyp-Enzyms führt es zu tiefgreifenden Veränderungen des Stoffwechsels von IDH1-mutierten Glioblastomen im Vergleich zu IDH1-Wildtyp-Glioblastomen oder gesunden Astrozyten. Unter anderem erhöht es die Abhängigkeit der Glioblastomzellen von Glutamin oder Glutamat als Energiequelle. Es wird angenommen, dass IDH1-mutierte Glioblastome einen sehr hohen Bedarf an Glutamat haben und diese Aminosäure und diesen Neurotransmitter als chemotaktisches Signal nutzen. Da gesunde Astrozyten Glutamat ausscheiden, bevorzugen IDH1-mutierte Glioblastomzellen keine dichten Tumorstrukturen, sondern wandern, dringen ein und verteilen sich in gesunden Teilen des Gehirns, wo die Glutamatkonzentration höher ist. Dies könnte eine Erklärung für das invasive Verhalten dieser IDH1-mutierten Glioblastome sein. ⓘ
Ionenkanäle
Darüber hinaus weist das GBM zahlreiche Veränderungen in Genen auf, die für Ionenkanäle kodieren, darunter eine Hochregulierung der gBK-Kaliumkanäle und der ClC-3-Chloridkanäle. Es wird angenommen, dass Glioblastom-Tumorzellen durch die Hochregulierung dieser Ionenkanäle eine verstärkte Ionenbewegung über die Zellmembran ermöglichen und dadurch die H2O-Bewegung durch Osmose erhöhen, was den Glioblastomzellen hilft, ihr Zellvolumen sehr schnell zu verändern. Dies ist hilfreich für ihr extrem aggressives invasives Verhalten, da schnelle Anpassungen des Zellvolumens die Bewegung durch die gewundene extrazelluläre Matrix des Gehirns erleichtern können. ⓘ
MicroRNA
Im Jahr 2012 wurde die RNA-Interferenz, in der Regel die microRNA, in Gewebekulturen, Pathologieproben und präklinischen Tiermodellen des Glioblastoms untersucht. Darüber hinaus deuten experimentelle Beobachtungen darauf hin, dass microRNA-451 ein wichtiger Regulator der LKB1/AMPK-Signalübertragung in kultivierten Gliomzellen ist und dass miRNA-Clustering epigenetische Signalwege bei der Krankheit steuert. ⓘ
Tumor-Gefäßsystem
Das GBM ist durch abnorme Gefäße gekennzeichnet, die eine gestörte Morphologie und Funktionalität aufweisen. Die hohe Permeabilität und schlechte Durchblutung des Gefäßsystems führen zu einem ungeordneten Blutfluss innerhalb des Tumors und können zu einer erhöhten Hypoxie führen, die wiederum das Fortschreiten des Krebses durch die Förderung von Prozessen wie der Immunsuppression begünstigt. ⓘ
Diagnose



Bei der Betrachtung mit MRT erscheinen Glioblastome häufig als ringförmige Läsionen mit Anreicherung. Dieses Erscheinungsbild ist jedoch nicht spezifisch, da auch andere Läsionen wie Abszesse, Metastasen, tumefaktive multiple Sklerose und andere Entitäten ein ähnliches Erscheinungsbild aufweisen können. Die endgültige Diagnose eines vermuteten GBM auf dem CT oder MRT erfordert eine stereotaktische Biopsie oder eine Kraniotomie mit Tumorresektion und pathologischer Bestätigung. Da der Tumorgrad auf dem bösartigsten Teil des Tumors basiert, kann eine Biopsie oder subtotale Tumorresektion zu einer Unterbewertung der Läsion führen. Die Darstellung des Tumordurchblutungsflusses mittels Perfusions-MRT und die Messung der Tumormetaboliten-Konzentration mittels MR-Spektroskopie können in ausgewählten Fällen den diagnostischen Wert der Standard-MRT erhöhen, indem sie ein erhöhtes relatives zerebrales Blutvolumen bzw. einen erhöhten Cholin-Peak anzeigen, aber die Pathologie bleibt der Goldstandard für die Diagnose und molekulare Charakterisierung. ⓘ
Die Unterscheidung zwischen primärem Glioblastom und sekundärem Glioblastom ist wichtig. Diese Tumoren entstehen spontan (de novo) oder haben sich aus einem Gliom niedrigeren Grades entwickelt. Primäre Glioblastome haben eine schlechtere Prognose, eine andere Tumorbiologie und sprechen unter Umständen anders auf die Therapie an, so dass dies eine kritische Bewertung ist, um Prognose und Therapie des Patienten zu bestimmen. Über 80 % der sekundären Glioblastome weisen eine Mutation in IDH1 auf, während diese Mutation beim primären Glioblastom selten ist (5-10 %). IDH1-Mutationen sind daher ein nützliches Instrument zur Unterscheidung zwischen primären und sekundären Glioblastomen, da sie histopathologisch sehr ähnlich sind und die Unterscheidung ohne molekulare Biomarker unzuverlässig ist. ⓘ

Histopathologie eines Glioblastoms, das die Merkmale eines hochgradigen Astrozytoms mit ausgeprägter nukleärer Pleomorphie, multiplen Mitosen (ein weißer Pfeil) und vielkernigen Zellen (ein schwarzer Pfeil) zeigt, wobei die Zellen in der H&E-Färbung eine musterlose Anordnung auf einem rosa fibrillären Hintergrund aufweisen.
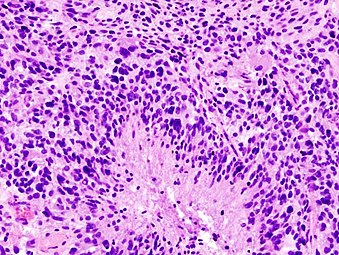
Die Histopathologie bei geringerer Vergrößerung zeigt eine Nekrose, die von Pseudopalisaden von Tumorzellen umgeben ist und die Diagnose eines Glioblastoms und nicht eines anaplastischen Astrozytoms zulässt. ⓘ

.jpg)
Die Diagnose wird zunächst durch bildgebende Verfahren wie Computertomographie (CT) oder Magnetresonanztomographie (MRT) gestützt. In der CT-Bildgebung mit Kontrastmittel erscheint das Glioblastom unregelmäßig geformt mit randständig starker Kontrastmittelaufnahme (ringförmiges Enhancement). Bei kleineren Tumoren ist dieses ringförmig konfiguriert, bei größeren bildet es eine girlandenartige Formation aus. In der Umgebung des Tumors bildet sich typischerweise ein erhebliches Ödem aus. Der MRT-Befund ist recht typisch: die soliden Anteile des Glioblastoms reichern Kontrastmittel stark an, dagegen heben sich die Aussparungen durch zystische Anteile und die Blutungen ab. Letztendlich wird die Diagnose am Tumorgewebe, das bei einer stereotaktischen Hirnbiopsie oder Tumorresektion gewonnen wurde, neuropathologisch bestätigt. Im Einzelfall werden Supplementäruntersuchungen wie Elektroenzephalografie und Lumbalpunktion durchgeführt, die der Einschätzung der Anfallsneigung bzw. der differentialdiagnostischen Abgrenzung gegen Hirnabszesse oder Lymphome dienen. ⓘ
Das Glioblastom geht von der weißen Substanz aus. Seine mit Abstand häufigste Lokalisation ist das Großhirn, wo es in allen Hirnlappen entstehen kann, aber den Frontal- und den Temporallappen bevorzugt. Im Bereich von Kleinhirn, Hirnstamm und Rückenmark sind Glioblastome selten. Oft wachsen hemisphärielle Glioblastome über den Balken auf die andere Seite hinüber. Solche Tumoren werden als sogenannte „Schmetterlingsgliome“ bezeichnet. Das Wachstum von Glioblastomen ist diffus infiltrierend. ⓘ
Vorbeugung
Es gibt keine bekannten Methoden zur Vorbeugung von Glioblastomen. Im Gegensatz zu einigen anderen Krebsarten treten die meisten Gliome ohne Vorwarnung auf, und es gibt keine bekannten Möglichkeiten, sie zu verhindern. ⓘ
Behandlung
Die Behandlung des Glioblastoms ist aufgrund mehrerer komplizierender Faktoren schwierig:
- Die Tumorzellen sind gegenüber herkömmlichen Therapien resistent.
- Das Gehirn ist anfällig für Schäden durch herkömmliche Therapien.
- Das Gehirn hat nur eine begrenzte Fähigkeit, sich selbst zu reparieren.
- Viele Medikamente können die Blut-Hirn-Schranke nicht überwinden, um auf den Tumor zu wirken. ⓘ
Die Behandlung von primären Hirntumoren besteht aus einer palliativen (symptomatischen) Behandlung und Therapien, die das Überleben verbessern sollen. ⓘ
Symptomatische Therapie
Die unterstützende Behandlung konzentriert sich auf die Linderung der Symptome und die Verbesserung der neurologischen Funktion des Patienten. Die wichtigsten unterstützenden Mittel sind Antikonvulsiva und Kortikosteroide.
- In der Vergangenheit wurden etwa 90 % der Patienten mit Glioblastom mit Antikonvulsiva behandelt, obwohl nur schätzungsweise 40 % der Patienten diese Behandlung benötigten. In jüngster Zeit wird den Neurochirurgen empfohlen, Antikonvulsiva nicht prophylaktisch zu verabreichen, sondern zu warten, bis ein Krampfanfall auftritt, bevor diese Medikamente verschrieben werden. Bei Patienten, die gleichzeitig mit Phenytoin bestrahlt werden, kann es zu schweren Hautreaktionen wie Erythema multiforme und Stevens-Johnson-Syndrom kommen.
- Kortikosteroide, in der Regel Dexamethason, können das peritumorale Ödem reduzieren (durch eine Umstrukturierung der Blut-Hirn-Schranke), den Masseneffekt verringern und den Hirndruck senken, wobei Kopfschmerzen oder Schläfrigkeit abnehmen. ⓘ
Chirurgie
Die Chirurgie ist die erste Stufe der Behandlung des Glioblastoms. Ein durchschnittlicher GBM-Tumor enthält 1011 Zellen, die nach der Operation auf durchschnittlich 109 Zellen reduziert sind (eine Reduktion um 99 %). Zu den Vorteilen eines chirurgischen Eingriffs gehören die Resektion für eine pathologische Diagnose, die Linderung von Symptomen im Zusammenhang mit dem Masseneffekt und die mögliche Entfernung der Krankheit, bevor eine sekundäre Resistenz gegen Strahlen- und Chemotherapie auftritt. ⓘ
Je größer das Ausmaß der Tumorentfernung ist, desto besser. Retrospektive Analysen haben gezeigt, dass die Entfernung von 98 % oder mehr des Tumors mit einer deutlich längeren Heilungsdauer verbunden ist als die Entfernung von weniger als 98 % des Tumors. Die Wahrscheinlichkeit einer nahezu vollständigen Entfernung des Tumors kann erhöht werden, wenn die Operation mit einem fluoreszierenden Farbstoff, der 5-Aminolävulinsäure, durchgeführt wird. GBM-Zellen sind zum Zeitpunkt der Diagnose im gesamten Gehirn weit verbreitet, und trotz einer "vollständigen Resektion" des gesamten offensichtlichen Tumors entwickeln die meisten Patienten mit GBM später entweder in der Nähe der ursprünglichen Stelle oder an weiter entfernten Stellen im Gehirn wiederkehrende Tumoren. Nach der Operation werden andere Behandlungsmethoden, in der Regel Strahlen- und Chemotherapie, eingesetzt, um das Wiederauftreten der Krankheit zu unterdrücken und zu verlangsamen. ⓘ
Strahlentherapie
Im Anschluss an die Operation ist die Strahlentherapie die wichtigste Behandlungsmethode für Menschen mit Glioblastom. Sie wird in der Regel zusammen mit der Verabreichung von Temozolomid durchgeführt. Eine entscheidende klinische Studie, die Anfang der 1970er Jahre durchgeführt wurde, zeigte, dass bei 303 GBM-Patienten, die nach dem Zufallsprinzip entweder bestrahlt oder nicht bestrahlt wurden, die mediane Überlebenszeit derjenigen, die bestrahlt wurden, mehr als doppelt so hoch war wie die derjenigen, die nicht bestrahlt wurden. Die nachfolgende klinische Forschung hat versucht, auf dem Grundgerüst der Operation mit anschließender Bestrahlung aufzubauen. Im Durchschnitt kann eine Strahlentherapie nach einer Operation die Tumorgröße auf 107 Zellen reduzieren. Die Ganzhirnbestrahlung bringt im Vergleich zur präziseren und gezielteren dreidimensionalen konformalen Strahlentherapie keine Verbesserung. Eine Gesamtstrahlendosis von 60-65 Gy hat sich als optimal für die Behandlung erwiesen. ⓘ
Es ist bekannt, dass GBM-Tumore Zonen mit hypoxischem Gewebe enthalten, die sehr resistent gegen eine Strahlentherapie sind. Es wurden verschiedene Ansätze für Chemotherapie-Radiosensibilisatoren verfolgt, mit begrenztem Erfolg (Stand 2016). Zu den neueren Forschungsansätzen gehören seit 2010 präklinische und klinische Untersuchungen zur Verwendung einer die Sauerstoffdiffusion fördernden Verbindung wie Transnatriumcrocetinat als Radiosensibilisator, und seit 2015 läuft eine klinische Studie. Die Borneutroneneinfangtherapie wurde als alternative Behandlungsmethode für Glioblastome getestet, wird aber nicht allgemein eingesetzt. ⓘ
Chemotherapie
Die meisten Studien zeigen keinen Nutzen einer zusätzlichen Chemotherapie. Eine große klinische Studie mit 575 Teilnehmern, die nach dem Zufallsprinzip einer Standardbestrahlung gegenüber einer Bestrahlung plus Temozolomid-Chemotherapie zugeteilt wurden, zeigte jedoch, dass die Gruppe, die Temozolomid erhielt, im Median 14,6 Monate überlebte, während die Gruppe, die nur bestrahlt wurde, 12,1 Monate überlebte. Dieses Behandlungsschema ist heute Standard für die meisten Fälle von Glioblastom, bei denen die Betroffenen nicht an einer klinischen Studie teilnehmen. Temozolomid scheint zu wirken, indem es die Tumorzellen für die Bestrahlung sensibilisiert, und scheint bei Tumoren mit MGMT-Promotor-Methylierung wirksamer zu sein. Hohe Dosen von Temozolomid bei hochgradigen Gliomen haben eine geringe Toxizität, aber die Ergebnisse sind mit den Standarddosen vergleichbar. Eine antiangiogene Therapie mit Medikamenten wie Bevacizumab lindert die Symptome, scheint aber das Gesamtüberleben von Glioblastom-Patienten nicht zu beeinflussen. Der Gesamtnutzen der antiangiogenen Therapien im Jahr 2019 ist unklar. Bei älteren Menschen mit neu diagnostiziertem Glioblastom, die einigermaßen fit sind, führt eine gleichzeitige und adjuvante Chemoradiotherapie zum besten Gesamtüberleben, ist aber mit einem höheren Risiko hämatologischer Nebenwirkungen verbunden als eine alleinige Radiotherapie. ⓘ
Cannabinoide
Die Wirksamkeit von Cannabinoiden (Cannabisderivaten) ist in der Onkologie bekannt (durch Kapseln mit Tetrahydrocannabinol (THC) oder dem synthetischen Analogon Nabilon), einerseits zur Bekämpfung der durch die Chemotherapie ausgelösten Übelkeit und des Erbrechens, andererseits zur Appetitanregung und zur Linderung des Angstgefühls oder der tatsächlichen Schmerzen. Ihre Fähigkeit, das Wachstum und die Angiogenese von bösartigen Gliomen in Mausmodellen zu hemmen, wurde nachgewiesen. Die Ergebnisse einer Pilotstudie über den Einsatz von THC bei Patienten mit rezidivierendem Glioblastom im Endstadium erscheinen einer weiteren Untersuchung würdig. Ein möglicher Weg für künftige Forschungen liegt in der Entdeckung, dass Cannabinoide in der Lage sind, die neoplastischen Stammzellen des Glioblastoms in Mausmodellen anzugreifen, um einerseits ihre Differenzierung in reifere, möglicherweise "behandelbarere" Zellen zu bewirken und andererseits die Tumorigenese zu hemmen. ⓘ
Andere Verfahren
Die elektrische Wechselfeldtherapie ist eine von der FDA zugelassene Therapie für neu diagnostizierte und wiederkehrende Glioblastome. Im Jahr 2015 berichteten erste Ergebnisse einer randomisierten klinischen Phase-III-Studie zur elektrischen Wechselfeldtherapie plus Temozolomid bei neu diagnostiziertem Glioblastom über eine dreimonatige Verbesserung des progressionsfreien Überlebens und eine fünfmonatige Verbesserung des Gesamtüberlebens im Vergleich zur alleinigen Temozolomid-Therapie, was die erste große Studie seit zehn Jahren darstellt, die eine Überlebensverbesserung in dieser Situation zeigt. Trotz dieser Ergebnisse ist die Wirksamkeit dieses Ansatzes unter medizinischen Experten nach wie vor umstritten. Das zunehmende Verständnis der mechanistischen Grundlagen, durch die die Therapie mit elektrischen Wechselfeldern krebshemmende Wirkungen entfaltet, und die Ergebnisse der laufenden klinischen Phase-III-Studien bei extrakraniellen Tumoren könnten jedoch dazu beitragen, dass die klinische Akzeptanz für die Behandlung des Glioblastoms in Zukunft zunimmt. ⓘ
Eine Studie der Universität Tel Aviv hat gezeigt, dass die pharmakologische und molekulare Hemmung des P-Selektin-Proteins in Mausmodellen des Glioblastoms zu einem geringeren Tumorwachstum und einer längeren Überlebensdauer führt. Die Ergebnisse dieser Forschung könnten zu möglichen Therapien mit Medikamenten führen, die dieses Protein hemmen, wie z. B. Crizanlizumab. ⓘ
Prognose
Die Überlebensdauer nach der Diagnose beträgt in der Regel 10 bis 13 Monate, wobei weniger als 1 bis 3 % der Betroffenen länger als fünf Jahre überleben. In den Vereinigten Staaten lag die Fünfjahresüberlebensrate zwischen 2012 und 2016 bei 6,8 %. Ohne Behandlung beträgt die Überlebensdauer in der Regel 3 Monate. Vollständige Heilungen sind extrem selten, wurden aber schon berichtet. ⓘ
Mit zunehmendem Alter (> 60 Jahre) steigt das prognostische Risiko. Der Tod ist in der Regel auf eine ausgedehnte Tumorinfiltration mit Hirnödem und erhöhtem intrakraniellen Druck zurückzuführen. ⓘ
Ein guter anfänglicher Karnofsky-Performance-Score (KPS) und MGMT-Methylierung sind mit einer längeren Überlebenszeit verbunden. Bei Glioblastomen kann ein DNA-Test durchgeführt werden, um festzustellen, ob der Promotor des MGMT-Gens methyliert ist oder nicht. Patienten mit einem methylierten MGMT-Promotor haben eine längere Überlebenszeit als Patienten mit einem nicht methylierten MGMT-Promotor, was zum Teil auf eine höhere Empfindlichkeit gegenüber Temozolomid zurückzuführen ist. Ein weiterer positiver prognostischer Marker für Glioblastom-Patienten ist die Mutation des IDH1-Gens, die mit DNA-basierten Methoden oder durch Immunhistochemie mit einem Antikörper gegen die häufigste Mutation, nämlich IDH1-R132H, getestet werden kann. ⓘ
Eine höhere prognostische Aussagekraft lässt sich durch die Kombination des Mutationsstatus von IDH1 und des Methylierungsstatus von MGMT zu einem Zwei-Gene-Prädiktor erzielen. Patienten mit sowohl IDH1-Mutationen als auch MGMT-Methylierung haben die längste Überlebenszeit, Patienten mit einer IDH1-Mutation oder MGMT-Methylierung eine mittlere Überlebenszeit und Patienten ohne eines der beiden genetischen Ereignisse haben die kürzeste Überlebenszeit. ⓘ
Langfristige Vorteile werden auch mit denjenigen Patienten in Verbindung gebracht, die eine Operation, Strahlentherapie und Temozolomid-Chemotherapie erhalten. Warum manche Patienten mit Glioblastom länger überleben, ist jedoch noch weitgehend unbekannt. Ein Alter unter 50 Jahren wird mit einer längeren Überlebenszeit bei GBM in Verbindung gebracht, ebenso wie eine Resektion von mehr als 98 %, die Anwendung einer Temozolomid-Chemotherapie und ein besserer KPS-Wert. Eine neuere Studie bestätigt, dass ein jüngeres Alter mit einer viel besseren Prognose verbunden ist, wobei ein kleiner Teil der Patienten unter 40 Jahren eine bevölkerungsbezogene Heilung erreicht. Man geht davon aus, dass eine Heilung eintritt, wenn das Sterberisiko einer Person auf das der Normalbevölkerung zurückgeht, und bei GBM ist dies nach 10 Jahren der Fall. ⓘ
Die UCLA Neuro-Onkologie veröffentlicht Echtzeit-Überlebensdaten für Patienten mit dieser Diagnose. ⓘ
Einer Studie aus dem Jahr 2003 zufolge lässt sich die GBM-Prognose in drei Untergruppen einteilen, die vom KPS, dem Alter des Patienten und der Behandlung abhängen. ⓘ
| Rekursive Partitionierungsanalyse (RPA) Klasse |
Definition | Historische mediane Überlebenszeit | Historische 1-Jahres-Überlebenszeit | Historische 3-Jahres-Überlebenszeit | Historische 5-Jahres-Überlebenszeit ⓘ |
|---|---|---|---|---|---|
| III | Alter < 50, KPS ≥ 90 | 17,1 Monate | 70% | 20% | 14% |
| IV | Alter < 50, KPS < 90 | 11,2 Monate | 46% | 7% | 4% |
| Alter ≥ 50, KPS ≥ 70, operative Entfernung bei guter neurologischer Funktion | |||||
| V + VI | Alter ≥ 50, KPS ≥ 70, chirurgische Entfernung mit schlechter neurologischer Funktion | 7,5 Monate | 28% | 1% | 0% |
| Alter ≥ 50, KPS ≥ 70, keine chirurgische Entfernung | |||||
| Alter ≥ 50, KPS < 70 |
Wegen der diffusen Infiltration des Hirngewebes durch Tumorzellen kommt es nach der Behandlung häufig innerhalb von Monaten zu einem Rezidiv. Einzelne Patienten können dessen ungeachtet mehrere Jahre bei relativ guter Gesundheit mit einem Glioblastom leben. Die Identifizierung klinischer und molekularer Faktoren, die charakteristisch für solche Langzeitüberlebenden sind, ist Gegenstand intensiver Forschung. ⓘ
Epidemiologie
Jährlich erkranken etwa drei von 100.000 Menschen an der Krankheit, wobei die regionale Häufigkeit wesentlich höher sein kann. Die Häufigkeit in England hat sich zwischen 1995 und 2015 verdoppelt. ⓘ
Es ist der zweithäufigste Krebs des zentralen Nervensystems nach dem Meningeom. Er tritt häufiger bei Männern als bei Frauen auf. Obwohl das Durchschnittsalter bei der Diagnose 64 Jahre beträgt, war 2014 die große Kategorie der Hirntumore bei den unter 20-Jährigen in den Vereinigten Staaten die zweithäufigste nach Leukämie. ⓘ
Historisches
Der Begriff Glioblastoma multiforme wurde 1926 von Percival Bailey und Harvey Cushing geprägt. Die Begriffsbildung basierte auf der Vorstellung, dass sich der Tumor aus primitiven Vorstufen von Gliazellen (Glioblasten) entwickelt, sowie der Beobachtung, dass das Erscheinungsbild mit Nekrosen, Einblutungen und Zysten sehr variabel (multiform) sein kann. Der von dem Pathologen Frank Burr Mallory bereits 1914 verwendete Begriff Spongioblastoma multiforme konnte sich nicht durchsetzen. ⓘ
Forschung
Gentherapie
Die Gentherapie wurde als Methode zur Behandlung des Glioblastoms erforscht. Während Tiermodelle und frühe klinische Studien erfolgreich waren, sind bis 2017 alle Gentherapie-Medikamente, die in klinischen Phase-III-Studien für das Glioblastom getestet wurden, gescheitert. Die Wissenschaftler haben die Kern-Schale-Nanostruktur LPLNP-PPT (Long Persistent Luminescence Nanoparticles) entwickelt. PPT steht für Polyetherimid, PEG und trans-Aktivator der Transkription, und TRAIL ist der humane tumor necrosis factor-related apoptosis-induced ligand) für eine effektive Genverabreichung und -verfolgung entwickelt, mit positiven Ergebnissen. Es handelt sich um einen TRAIL-Liganden, der kodiert wurde, um die Apoptose von Krebszellen, insbesondere von Glioblastomen, auszulösen. Obwohl sich diese Studie 2017 noch in der klinischen Erprobung befand, hat sie diagnostische und therapeutische Funktionalitäten gezeigt und wird für klinische Anwendungen in der stammzellbasierten Therapie von großem Interesse sein. ⓘ
Onkolytische Virotherapie
Die onkolytische Virotherapie ist eine neuartige Behandlungsmethode, die sowohl in präklinischen als auch in klinischen Phasen untersucht wird. Mehrere Viren, darunter Herpes-Simplex-Viren, Adenoviren, Polioviren und Reoviren, werden derzeit in den Phasen I und II der klinischen Versuche zur Glioblastomtherapie getestet und haben gezeigt, dass sie das Gesamtüberleben verbessern. ⓘ
Intranasale Verabreichung von Medikamenten
Die direkte Verabreichung von Arzneimitteln über die Nase an das Gehirn wird als Mittel zur Erzielung höherer und hoffentlich wirksamerer Arzneimittelkonzentrationen im Gehirn erforscht. In einer klinischen Phase-I/II-Studie mit Glioblastom-Patienten in Brasilien wurde der Naturstoff Perillylalkohol für die intranasale Verabreichung als Aerosol untersucht. Die Ergebnisse waren ermutigend, und 2016 wurde eine ähnliche Studie in den Vereinigten Staaten eingeleitet. ⓘ
Klinische Erscheinungen
Wegen des raschen Wachstums entwickeln sich die Beschwerden meistens rasch innerhalb weniger Wochen bis Monate. Erste Symptome können anhaltende und ungewohnte Kopfschmerzen, aber auch neu auftretende epileptische Anfälle sein. Fokale neurologische Ausfälle wie Lähmungen, Aphasien und Sehstörungen können lokalisationsabhängig hinzukommen. Schließlich sind es oft auffällige Persönlichkeitsveränderungen, Apathie oder psychomotorische Verlangsamung, die den Patienten zum Arzt führen. Hirndruckzeichen wie Stauungspapille, Erbrechen, Somnolenz und Koma treten spät auf und sind prognostisch ungünstig. ⓘ
Pathologie

Das Glioblastom ist durch seine inhomogene und vielfältige (daher: multiforme) Erscheinung gekennzeichnet: die Tumorschnittfläche weist häufig rötliche Einblutungen und gelbliche Gewebsuntergänge (Nekrosen) auf. ⓘ
Histologie
.jpg)
Feingeweblich (histologisch) handelt es sich um zelldichte astrozytär differenzierte Tumoren, die diffus das umgebende reaktiv veränderte Hirngewebe infiltrieren. Die Tumorzellen sind mit multipolaren feinen Fortsätzen fibrillär-astrozytär differenziert oder weisen mit einem aufgeblähten Zytoplasma eine gemästet-zellige Differenzierung auf. Auch Riesenzellen mit bizarren Kernen oder kleinzellige Areale mit wenig ausgedehnten Zellkörpern kommen vor. Die Zellkerne sind meist chromatinreich und vielgestaltig (polymorph). Mitotische und proliferative Aktivität sind erhöht. ⓘ
Entscheidend für die Diagnose des Glioblastoms (und die Abgrenzung gegenüber dem anaplastischen Astrozytom) ist nach der WHO-Klassifikation der Tumoren des zentralen Nervensystems jedoch der Nachweis von Tumornekrosen (flächenhaft oder typischerweise strichförmig mit perifokaler Zelldichtesteigerung) oder hochgradig pathologischer Blutgefäße. ⓘ
Varianten
Bei Gliosarkomen handelt es sich um Glioblastome, die neben den oben beschriebenen astrozytären Tumoranteilen auch bindegewebsreiche sarkomatöse Abschnitte mit spindelzelligen Tumorzellen aufweisen. Ein Epitheloides Glioblastom weist große Epitheloidzellen mit reichlich eosinophilem Zytoplasma auf. Als Riesenzellglioblastome werden Glioblastome mit einer ausgeprägten riesenzelligen Komponente bezeichnet. Ebenfalls abzugrenzen sind Glioblastome mit oligodendroglialer Komponente, die möglicherweise eine etwas günstigere Prognose haben. ⓘ
Immunhistochemie

Immunhistochemisch ist in den Tumorzellen – wie in denen anderer glialer Hirntumoren – das saure Gliafaserprotein (glial fibrillary acidic protein, GFAP) nachweisbar, was in den meisten Fällen die Abgrenzung gegenüber Hirnmetastasen erlaubt. ⓘ