Liposarkom
| Liposarkom ⓘ | |
|---|---|
 | |
| Histopathologie eines Liposarkoms, H&E-Färbung: - | |
| Fachgebiet | Dermatologie, Allgemeinchirurgie, Onkologie |
| Symptome | Knoten unter der Haut, Schmerzen, Schwellung, Organfunktionsstörungen |
Liposarkome sind die häufigste Unterart der Weichteilsarkome und machen mindestens 20 % aller Sarkome bei Erwachsenen aus. Weichteilsarkome sind seltene Neoplasien mit über 150 verschiedenen histologischen Subtypen oder Formen. Liposarkome entstehen aus den Vorläuferlipoblasten der Adipozyten (d. h. Fettzellen) in den Fettgeweben. Fettgewebe ist über den ganzen Körper verteilt, unter anderem in den tiefen und oberflächlichen Schichten des Unterhautgewebes sowie in chirurgisch weniger zugänglichen Bereichen wie dem Retroperitoneum (d. h. dem Raum hinter der Bauchhöhle) und dem viszeralen Fett innerhalb der Bauchhöhle. ⓘ
Alle Liposarkome bestehen zumindest aus einigen Zellen, die bei der histopathologischen Untersuchung unter dem Mikroskop Ähnlichkeit mit Fettzellen aufweisen. Es gibt jedoch verschiedene Formen von Liposarkomen, die sich in ihrem klinischen Erscheinungsbild (z. B. Alter, Geschlecht, Lage des Tumors, Anzeichen und Symptome), ihrem Schweregrad (d. h. Potenzial, in lokales Gewebe einzudringen, nach chirurgischer Entfernung wieder aufzutreten und in entferntes Gewebe zu metastasieren), ihren genetischen Anomalien, ihrer Prognose und ihren bevorzugten Behandlungsmethoden unterscheiden. Die Weltgesundheitsorganisation hat die Liposarkome im Jahr 2020 in fünf mehr oder weniger ausgeprägte Formen eingeteilt: 1) atypischer lipomatöser Tumor/gut differenziertes Liposarkom; 2) entdifferenziertes Liposarkom; 3) myxoides Liposarkom; 4) pleomorphes Liposarkom und 5) myxoides pleomorphes Liposarkom. (Pleomorph weist auf das Vorhandensein von Zellen hin, die abnormale und oft große Variationen in ihrer Größe und Form und/oder der Größe und Form ihrer Zellkerne aufweisen). ⓘ
Während Liposarkomformen als aggressiv und bösartig oder, im Falle des atypischen lipomatösen Tumors/ gut differenzierten Liposarkoms, als relativ nicht aggressiv und gutartig eingestuft werden, können alle fünf Liposarkomformen lokal infiltrieren und nahe gelegene Gewebe und Organe verletzen, an chirurgisch unzugänglichen Stellen in der Nähe lebenswichtiger Organe (z. B. im Retroperitoneum) auftreten, nach chirurgischer Entfernung wiederkehren und sich zu lebensbedrohlichen Krankheiten entwickeln. Bisherige Studien haben ergeben, dass alle fünf Liposarkomformen zwar in der Regel zumindest anfänglich durch chirurgische Resektion behandelbar sind, aber oft nur geringfügig auf die derzeit verwendeten Chemo- und Strahlentherapien ansprechen. Die Liposarkome bedürfen einer Vielzahl weiterer Studien, um zu ermitteln, wie gut sie auf verschiedene Strahlen- und Chemotherapien sowie auf neuartige Behandlungsmethoden ansprechen, die einzeln und in verschiedenen Kombinationen angewandt werden und, wenn möglich, eine chirurgische Entfernung einschließen. ⓘ
| Klassifikation nach ICD-10 ⓘ | |
|---|---|
| C49.- | Bösartige Neubildung sonstigen Bindegewebes und anderer Weichteilgewebe |
| ICD-10 online (WHO-Version 2019) | |
| Klassifikation nach ICD-O-3 ⓘ | |
|---|---|
| 8850/3 | Liposarkom o.n.A. |
| 8851/3 | Gut differenziertes Liposarkom |
| 8852/3 | Myxoides Liposarkom |
| 8853/3 | Rundzelliges Liposarkom |
| 8854/3 | Pleomorphes Liposarkom |
| 8855/3 | Gemischtzelliges Liposarkom |
| ICD-O-3 erste Revision online | |






Das Liposarkom ist ein seltener bösartiger Tumor des Weichteilgewebes (Sarkom), der feingewebliche Merkmale von Fettzellen oder Fettzellvorstufen aufweist. Mit einem Anteil von 16–18 % ist das Liposarkom nach dem malignen fibrösen Histiozytom das zweithäufigste Weichteilsarkom. Die Erstbeschreibung des Liposarkoms als Krankheitsentität erfolgte 1857 durch Rudolf Virchow. ⓘ
Formen von Liposarkomen
Liposarkome sind im Allgemeinen große Tumore (>10 cm), können aber fast jede Größe haben. Sie treten hauptsächlich bei Erwachsenen auf, wobei nur 0,7 % der Fälle bei Personen unter 16 Jahren auftreten. Bei Erwachsenen treten Liposarkome vor allem im und nach dem mittleren Lebensalter auf. Bei den sehr seltenen Fällen, die bei Kindern und Jugendlichen auftreten, wird überwiegend die Form des myxoiden Liposarkoms diagnostiziert. ⓘ
Die fünf Liposarkomformen müssen nicht nur voneinander, sondern auch von bestimmten anderen Weichteiltumoren unterschieden werden. Diese anderen Tumoren und einige ihrer charakteristischen histopathologischen Merkmale sind: 1) dysplastische Lipome (d. h. gutartige Humore mit Gewebsnekrosen und neoplastischen Fettzellen unterschiedlicher Größe mit unterschiedlich großen/geformten Kernen; diese neoplastischen Zellen überexprimieren im Gegensatz zu den meisten neoplastischen Zellen der Liposarkome das MDM2-Gen nicht); 2) atypische spindelzellige Lipome (d. h. d. h. gutartige Tumore mit leicht atypischen spindelförmigen Zellen in einem faserigen bis myxoiden Stroma, durchsetzt mit vakuolierten Lipoblasten und Adipozyten unterschiedlicher Größe mit atypischen Kernen; 3) pleomorphe Lipome (d. h. d. h. gutartige Tumore, die durch Riesenzellen mit überlappenden Kernen gekennzeichnet sind); und 4) solitäre fibröse Tumore (d. h. Tumore, von denen bis zu 22 % ein bösartiges Verhalten aufweisen, bestehend aus spindel- oder eiförmigen Zellen in einem kollagenen Hintergrundstroma, das mit Blutgefäßen mit einer charakteristischen Hirschhornform durchsetzt ist). ⓘ
Atypischer lipomatöser Tumor/gut differenziertes Liposarkom
Atypische lipomatöse Tumoren (ALT) und gut differenzierte Liposarkome (WDL) machen zusammen 40 bis 45 % aller Liposarkome aus. Sie metastasieren selten oder nie und gelten daher als gutartige oder prämaligne Tumoren. Sie sind jedoch lokal invasiv und können sich in ein aggressiveres und potenziell metastasierendes Liposarkom, d. h. ein dedifferenziertes Liposarkom, verwandeln. Außerdem kann ein chirurgisch entfernter atypischer lipomatöser Tumor bzw. ein gut differenziertes Liposarkom als dedifferenziertes Liposarkom wiederkehren. ⓘ
Präsentation

ALTs und WDLs gelten als praktisch identische Tumoren, mit der Ausnahme, dass ALTs definitionsgemäß Tumoren bezeichnen, die sich in den Armen oder Beinen entwickeln, während WDLs Tumoren bezeichnen, die sich an weniger gut chirurgisch zugänglichen Stellen entwickeln, z. B. in den tiefen, zentral gelegenen Weichteilen des Retroperitoneums, der paratestikulären Region (d. h. dem Bereich innerhalb des Hodensacks, der die Hoden, den Samenstrang, die Hodentunika, die Nebenhoden und den Hodenfortsatz umfasst), der Mundhöhle und der Augenhöhle. Diese Terminologie hat prognostische Auswirkungen: weniger als 7 % der ALT-Tumoren wandeln sich innerhalb von durchschnittlich 7 Jahren in dedifferenzierte Liposarkome um, während sich 17 % der WDL-Tumoren innerhalb von durchschnittlich 8 Jahren in dieses bösartigere Liposarkom umwandeln. ALT- und WDL-Tumore (im Folgenden ALT/WDL genannt) treten typischerweise bei Personen mittleren und höheren Alters als langsam wachsende Massen auf, die tendenziell größer und in einem fortgeschritteneren Stadium sind, wenn sie in tiefem Gewebe liegen. Diese Tumoren sind in der Regel nicht schmerzhaft und, wenn sie oberflächlich lokalisiert sind, leicht zu erkennen; sie können in den betroffenen Bereichen, wie z. B. dem Oberschenkel (siehe nebenstehende Abbildung), aufgrund ihres Eindringens in die Blut- und/oder Lymphgefäße, die den Tumorbereich entwässern, auch umfangreiche Ödeme (d. h. Schwellungen aufgrund lokaler Flüssigkeitsansammlungen) verursachen. Tief sitzende ALT/WDL-Tumoren können asymptomatisch sein, aber je nach Lage schwere Anzeichen und/oder Symptome einer Funktionsstörung in einem der verschiedenen Organe hervorrufen, die sie infiltrieren. Zu diesen Organen gehören die Organe in der Nähe oder im Retroperitoneum (z. B. Darm, Niere und die Harnleiter der Niere), die paratestikuläre Region, das Mediastinum (z. B. Luftröhre und die Hauptbronchien der Lunge) und der Kopf (z. B. der retrobulbäre Raum hinter dem Augapfel). ⓘ
Pathologie

Histopathologisch werden ALT/WDL-Tumore in adipozytäre/lipomähnliche, sklerosierende und entzündliche Varianten unterteilt, wobei adipozytäre/lipomähnliche Varianten am häufigsten vorkommen. Adipozytäre/lipomähnliche ALT/WDL-Tumoren bestehen aus Läppchen reifer Fettzellen, die von unregelmäßigen fibrösen Septen durchzogen sind (siehe nebenstehende H&E-gefärbte Mikroaufnahme). Sklerosierende ALT/WDL-Tumoren, die zweithäufigste Variante, entwickeln sich vor allem im retroperitonealen und paratestikulären Bereich; sie bestehen aus verstreuten, atypischen Stromazellen innerhalb eines kollagenen (d. h. kollagenhaltigen) Stromagewebes. Seltene vakuolenhaltige Lipoblasten bevölkern dieses Gewebe. Entzündliche ALT/WDL-Tumoren sind die seltenste Variante. Sie treten am häufigsten im Retroperitoneum auf und bestehen aus chronischen Entzündungszellen, z. B. Lymphozyten und Plasmazellen, sowie gelegentlichen lymphknotenähnlichen Follikeln, die in einem fettzellhaltigen Gewebehintergrund verstreut sind. ⓘ
Genetik
Die neoplastischen Zellen in ALT/WDL-Tumoren enthalten ein oder mehrere zusätzliche ringförmige kleine überzählige Markerchromosomen (sSMC) oder ein abnormales Riesenmarkerchromosom (d. h. ein ehemals normales Chromosom, das durch eine Verdopplung von Teilen seines eigenen oder eines oder mehrerer anderer Chromosomen abnormal geworden ist). Diese abnormalen Chromosomen enthalten zusätzliche Kopien des langen Arms von Chromosom 12 (auch q-Arm genannt) an den Bändern 13 bis 15. Dieser Abschnitt von Chromosom 12 enthält das Proto-Onkogen MDM2 (ein potenziell tumorauslösendes Gen, wenn es überexprimiert wird) in Band 15 und CDK4 (ein Gen, das bei Überexpression die Entwicklung verschiedener Tumore fördert) in Band 14.1. Die Amplifikation (d. h. erhöhte Kopien eines Gens ohne proportionalen Anstieg anderer Gene) dieser beiden Gene ist ein hochempfindlicher und spezifischer Indikator dafür, dass es sich bei einem Liposarkom entweder um ein ALT/WDL oder ein dedifferenziertes Liposarkom und nicht um eine andere Liposarkom- oder Lipomform handelt. Neben den Genen MDM2 und CDK4 enthält dieser Chromosomenbereich 13-15 auch die Gene TSPAN31 und HMGA2, die, wenn sie überexprimiert sind, mit verschiedenen Tumoren und/oder Krebsarten in Verbindung gebracht werden. Es wurde vermutet, dass eines oder mehrere dieser überexprimierten Gene die Entwicklung und/oder das Fortschreiten von ALT/WDL-Tumoren fördern und/oder dazu beitragen. ⓘ
Diagnose
Die Diagnose von ALT/WDL-Tumoren wird auf der Grundlage des klinischen Erscheinungsbildes, der Histopathologie und der genetischen Befunde gestellt. Insbesondere der Nachweis eines überexprimierten MDM2- oder CDK4-Gens in den ALT/WDL-Tumorzellen oder das Vorhandensein entweder des spezifischen ALT/WDL-assoziierten sSMC- oder Riesenmarker-Chromosoms (wie durch DNA-Sequenzierung der nächsten Generation, vergleichende genomische Hybridisierung und/oder hochspezialisierte zytogenetische G-Banding-Analysen definiert) stützt die Diagnose ALT/WDL oder dedifferenziertes Liposarkom nachdrücklich. Die klinische Präsentation und die histopathologischen Unterschiede zwischen den beiden letztgenannten Liposarkomformen helfen in der Regel bei der Unterscheidung zwischen ihnen. ⓘ
Behandlung und Prognose
ALT/WDL-Tumoren werden durch eine radikale chirurgische Resektion behandelt, bei der das gesamte neoplastische Tumorgewebe entfernt wird. Allerdings treten diese Tumore in 30-50 % der Fälle lokal wieder auf. Rezidive treten am häufigsten bei Tumoren auf, die sich an weniger gut zugänglichen Stellen befinden, z. B. im Retroperitoneum, im Mediastinum und im Samenstrang. Diese chirurgisch weniger gut zugänglichen Tumoren neigen dazu, wiederholt zu rezidivieren und können aufgrund ihrer schädigenden Wirkung auf lebenswichtige Organe letztlich zum Tod führen. Während ALT/WDL-Tumoren ein sehr geringes Metastasierungspotenzial haben, wandeln sich etwa 10 % in eine offen bösartige und potenziell metastasierende Liposarkomform, das dedifferenzierte Liposarkom, um. Die mediane Zeit für diese bösartige Umwandlung beträgt etwa 7-9 Jahre. Darüber hinaus kann ein chirurgisch entferntes ALT/WDL nach einem variablen Intervall als dedifferenziertes Liposarkom wieder auftreten. Eine große randomisierte, kontrollierte Studie, in der bei ALT/WDL-Tumoren eine Strahlentherapie mit anschließender Operation mit einer alleinigen Operation verglichen wurde, ergab kaum einen Unterschied zwischen den beiden Therapien. Kleinere Studien, in denen selektive Inhibitoren der Proteinprodukte der CDK4- oder MDM2-Gene, die bei ALT/WDL eine Rolle spielen, eingesetzt wurden, haben bestenfalls bescheidene Auswirkungen gezeigt. Weitere Studien, in denen diese oder völlig neue Behandlungsschemata eingesetzt werden, werden derzeit untersucht. In einer Übersichtsstudie aus dem Jahr 2012 wurden die 5- und 10-Jahres-Überlebensraten von Personen mit ALT/WDL mit 100 % bzw. 87 % angegeben. ⓘ
Neuartige Therapien
Die neuartigen Therapien für ALT/WDL sind dieselben wie die, die im Abschnitt über neuartige Therapien für dendifferenzierte Liposarkome aufgeführt sind. ⓘ
Entdifferenziertes Liposarkom
Dedifferenzierte Liposarkome sind bösartige Tumore, die sich in ~10 % der Fälle aus einem bestehenden atypischen lipomatösen Tumor/gut differenzierten Liposarkom (ALT/WDL) oder an der Stelle entwickeln, an der ein ALT/WPL-Tumor operativ entfernt wurde. Bei Personen mit einer De-novo-Diagnose dieses Tumors kann ein ALT/WDL-Tumor zu einem dedifferenzierten Liposarkom fortgeschritten sein, das jedoch unentdeckt blieb, weil es sich asymptomatisch an einer stark abgeschirmten Stelle wie dem Retroperitoneum oder der Bauchhöhle entwickelte. Viele der klinischen und genetischen Merkmale der dedifferenzierten Liposarkome ähneln denen von ALT/WDL-Tumoren. ⓘ
Präsentation
Dedifferenzierte Liposarkome (DDL) treten am häufigsten bei Erwachsenen mittleren Alters und älteren Menschen auf, mit einem Häufigkeitsgipfel im sechsten bis achten Lebensjahrzehnt. Selten sind diese Tumoren auch bei Kindern und Jugendlichen aufgetreten. DDL-Tumoren treten am häufigsten im Retroperitonealraum auf, können aber ähnlich wie ALT/WDL auch in den Extremitäten, im paratestikulären Bereich, im Mediastinum, im Kopf oder am Hals vorkommen. Weniger als 1 % aller DDLs entwickeln sich in oberflächlichen Weichteilen oder der Augenhöhle. Bei der Vorstellung sind DDL-Tumoren in der Regel schmerzlos, groß, können sich seit Jahren langsam und progressiv vergrößern und weisen auf Routineröntgenbildern Bereiche mit Kalkablagerungen auf (siehe Abb. 1 im Abschnitt Histopathologie von Liposarkomen). Seltener treten bei den Betroffenen Anzeichen und/oder Symptome auf, die darauf zurückzuführen sind, dass der Tumor auf ein Organ drückt (z. B. Bauchschmerzen durch Verstopfung des Darms oder Harnwegsobstruktion durch Verstopfung der Harnröhre). Sehr selten zeigen Personen mit DDL ein oder mehrere Anzeichen oder Symptome einer chronischen Entzündung (siehe B-Symptome) und/oder eines der endrokrinen, neurologischen, mukokutanen, hämatologischen oder anderen gewebebezogenen paraneoplastischen Syndrome. Die Anzeichen und Symptome der chronischen Entzündung und der verschiedenen paraneoplastischen Syndrome werden durch die Sekretion von Zytokinen, Hormonen, Prostaglandinen und/oder anderen systemisch wirkenden Stoffen durch den Tumor verursacht; sie verschwinden nach erfolgreicher Behandlung der DDL vollständig. ⓘ
Pathologie
Das histopathologische Erscheinungsbild von DDL-Tumoren (siehe Abb. 2 im Abschnitt Histopathologie von Liposarkomen) ist sehr unterschiedlich, weist aber am häufigsten Merkmale undifferenzierter pleomorpher Sarkome (das sind Tumore, die dicht mit Zellen unterschiedlicher Größe und Form bevölkert sind und Kerne unterschiedlicher Größe und Form enthalten) oder spindelzelliger Sarkome (das sind Tumore, die aus spindelförmigen Zellen auf bindegewebigem Hintergrund bestehen) auf. Verschiedene Teile von DDL-Tumoren zeigen oft Variationen im Erscheinungsbild ihres Bindegewebshintergrunds: Diese Gewebe können myxoid (d. h. aus einer klaren, schleimartigen Substanz bestehend, die bei der Färbung mit einer Standard-H&E-Färbemethode blauer oder violetter erscheint als die rote Farbe von normalem Gewebe) oder myxokollagen (d. h. hoher Kollagenfasergehalt im Gewebe) sein. d. h. hoher Gehalt an Kollagenfasern auf myxoidem Hintergrund), und in ~5 % der Fälle finden sich Bereiche mit osteoidem (siehe Abb. 1 im Abschnitt Histopathologie von Liposarkomen) oder knorpeligem Material. Die Tumoren weisen auch große Unterschiede in ihrem Zellinhalt auf. So weisen beispielsweise bis zu 10 % der DDL-Tumoren Bereiche mit ALT/WDL-Histopathologie auf, und seltene Fälle von DDL haben Bereiche, die meningothelartige Wirbel flacher Zellen enthalten. ⓘ
Genetik
Die neoplastischen Zellen in DDL und ALT/WDL tragen ähnliche kleine überzählige Markerchromosomen (sSMCs) und/oder riesige Markerchromosomen, die zusätzliche Teile des q-Arms von Chromosom 12 in den Bändern 13 bis 15 enthalten. In diesem chromosomalen Bereich befinden sich zwei Gene, die mit der Tumorentwicklung in Verbindung gebracht werden, nämlich die Gene MDM2 und CDK4. Das Vorhandensein zusätzlicher Kopien dieser beiden Gene und/oder ihrer überproduzierten Proteinprodukte ist ein hochempfindlicher und spezifischer Indikator dafür, dass es sich bei einem lipomatösen Tumor um einen ALT/WDL- oder DDL-Tumor handelt und nicht um eine andere Art von lipomatösem Tumor. Die Überexpression der MDM2- und CDK-Gene und/oder anderer genetischer Stoffe in den sSMCs oder Riesenmarker-Chromosomen steht im Verdacht, die Entwicklung und/oder das Fortschreiten von DDL- sowie ALT/WDL-Tumoren zu fördern. Zu den anderen Genen im sMMC- und Riesenmarker-Chromosom, die auch in neoplastischen ALT/WDL- und DDL-Zellen überexprimiert sind, gehören HMGA2, CPM, YEATS4 und DDIT3. Im Vergleich zu den neoplastischen ALT/WDL-Zellen exprimieren die neoplastischen DDL-Zellen jedoch 1) mehr Gene auf den beiden abnormalen Chromosomen, was zur Progression von ALT/WDL zu DDL beitragen kann, und 2) mehr Genprodukte auf dem langen Arm von Chromosom 1 bei Band 32, dem langen Arm von Chromosom 6 bei Band 33 und in ~25 % der Fälle auf dem kurzen Arm von Chromosom 1 bei Band 32. 2, der das JUN-Gen enthält (dieses Gen ist bei DDL überexprimiert, nicht aber bei ALT/WDL). Da das Produkt des JUN-Gens, c-jun, den Zelltod hemmt und die Zellproliferation fördert, kann seine Überproduktion zum Fortschreiten von ALT/WDL zu DDL und/oder zur Bösartigkeit der neoplastischen Zellen der DDL beitragen. Die Erstellung von Genexpressionsprofilen (d. h. die Messung der Expression der Produkte von Tausenden von Genen, die von Zellen, Geweben oder Tumoren gebildet werden) hat gezeigt, dass die Differenzierung von Fettzellen und Stoffwechselwege bei ALT/WDL hochreguliert sind, während die Zellproliferation und die DNA-Schadensreaktionswege bei DDL hochreguliert sind. ⓘ
Diagnose
Die Histopathologie der DDL ist oft nicht eindeutig genug, um eine sichere Diagnose zu stellen. Die Diagnose einer DDL wird jedoch bei Personen gestellt, deren Tumoren ALT/WDL mit histologischen DDL-Komponenten vermischt sind, die in der Vergangenheit bereits eine ALT/WDL hatten oder bei denen ein retroperitoneales Liposarkom vorliegt (DDL machen ~57 % aller retroperitonealen Liposarkome aus). DDL-Tumoren treten nur selten (<1 % der Fälle) als oberflächliche Hauttumoren auf, sind fast fünfmal seltener als ALT/WDL in der Augenhöhle zu finden und sind bei Kindern extrem selten. Der Nachweis einer MDM2-Amplifikation in den Tumorzellen ist der diagnostische Goldstandard, um WDL von Lipomen, dysplastischen Lipomen, atypischen spindelzelligen Sarkomen, pleomorphen Lipomen und solitären fibrösen Tumoren zu unterscheiden. Alternativ unterstützt der Nachweis eines überexprimierten CDK4-Gens in den Tumorzellen oder das Vorhandensein entweder der spezifischen ALT/WDL-assoziierten sSMCs oder des Riesenmarkerchromosoms die Diagnose DDL oder ALT/WDL. Die klinische Präsentation, die Histopathologie und die Genunterschiede (z. B. die Überexpression des cJUN-Gens in den Tumorzellen begünstigt die Diagnose DDL gegenüber ATL/WDL) zwischen den beiden letztgenannten Liposarkomformen helfen in der Regel bei der Unterscheidung zwischen ihnen. ⓘ
Behandlung und Prognose
Die vollständige chirurgische Resektion ist in der Regel die empfohlene Erstlinienbehandlung für lokalisierte DDL-Tumoren. Neue Studien deuten jedoch darauf hin, dass Patienten mit DDL-Tumoren, die auf eine Extremität oder den Rumpf beschränkt sind und eine prognostizierte 10-Jahres-Gesamtüberlebensrate von 51 % oder weniger haben, bessere Ergebnisse erzielen, wenn ihre chirurgische Behandlung durch eine Chemotherapie (z. B. Doxorubicin plus Ifosfamid) ergänzt wird. Bei diesen lokalisierten Formen der DDL kann auch eine perioperative Strahlentherapie gemäß den Leitlinien des National Comprehensive Cancer Network in Betracht gezogen werden. ⓘ
Die retroperitoneale DDL ist die häufigste, chirurgisch nicht zu behandelnde und schwerwiegende Form der DDL: Sie hat eine Rezidivrate von 66 % und eine Fünfjahres-Gesamtüberlebensrate von 54 %. Die wichtigste Behandlungsoption für die retroperitoneale DDL ist die chirurgische Resektion. In einer klinischen Phase-III-Studie wurde festgestellt, dass die Ergebnisse einer Strahlentherapie mit anschließender chirurgischer Resektion im Vergleich zur alleinigen chirurgischen Resektion bei der Behandlung der retroperitonealen DDL kaum Unterschiede aufweisen. In anderen klinischen Phase-III-Studien wurden DDL-Patienten mit unzugänglichen retroperitonealen und/oder metastasierten Tumoren mit einer Erstlinien-Chemotherapie behandelt, wobei Doxorubicin mit Doxorubicin plus Ifosfamid oder Doxorubicin mit Gemcitabin plus Docetaxel verglichen wurde. Andere Studien haben ebenfalls den Wert verschiedener Chemotherapieschemata untersucht. In diesen Studien wurden oft nur geringe Unterschiede in der Gesamtüberlebenszeit festgestellt, aber Verbesserungen beim progressionsfreien Überleben und anderen klinischen Parametern. Auf der Grundlage dieser Studien wird als Erstlinientherapie für retroperitoneale und andere chirurgisch nicht behandelbare oder metastasierte DDL-Tumoren eine Behandlung mit einem Anthrazyklin-basierten Chemotherapieschema oder, in tumorresistenten oder rezidivierenden Fällen, eine Eribulin-Chemotherapie empfohlen. In einer Übersichtsarbeit aus dem Jahr 2020 wurde die mediane Überlebenszeit für DDL mit niedrigem histopathologischem Grad auf 113 Monate und für DDL mit hohem histopathologischem Grad auf 48 Monate festgelegt. Weitere Studien sind erforderlich, um die Wirksamkeit von Strahlentherapie, Chemotherapie und neuartigen Therapien bei allen Formen der DDL zu belegen. ⓘ
Neuartige Therapien
Mehrere neue Therapieschemata für DDL und die aggressiveren oder anderweitig problematischen Fälle von ALT/WDL werden derzeit in klinischen Studien erprobt. Derzeit läuft eine klinische Studie der Phase II zu Abemaciclib bei Patienten mit vorbehandelter oder unbehandelter DDL. Vorläufige Analysen zeigten, dass dieser Inhibitor der Serin/Threonin-spezifischen Proteinkinase-Enzyme der CDK4- und CDK6-Gene zu einer Verlängerung der medianen progressionsfreien Überlebenszeit von 30,4 Wochen führte. Eine multizentrische, randomisierte, doppelblinde, placebokontrollierte klinische Studie der Phase III zu Abemaciclib befindet sich in der aktiven Phase und wird demnächst (wie im Juli 2021 angekündigt) mit der Rekrutierung von 108 Personen mit fortgeschrittener, rezidivierender und/oder metastasierter DDL beginnen. Die Studie wird von der Sarcoma Alliance for Research through Collaboration in Zusammenarbeit mit Eli Lilly and Company gesponsert. Ribociclib, ebenfalls ein CDK4- und CDK6-Geninhibitor, befindet sich in Kombination mit dem mTOR-Inhibitor Everolimus in einer klinischen Studie der Phase II bei Patienten mit fortgeschrittenem DDL oder Leiomyosarkom. In einer Phase-III-Zulassungsstudie (d. h. einer großen Bestätigungsstudie, die ein akzeptables Nutzen-/Sicherheitsprofil erstellen soll, um die Zulassung für eine genau definierte Indikation zu erhalten) werden die Sicherheit und Wirksamkeit von Milademetan im Vergleich zu Trabectedin bei Patienten mit inoperablem (d. h. eine Resektion wird als inakzeptable Morbidität oder Mortalität angesehen) oder metastasiertem DDL untersucht, das unter einer oder mehreren früheren systemischen Therapien, einschließlich mindestens einer Anthrazyklin-basierten Therapie, fortgeschritten ist. Der Sponsor, Rain Therapeutics Inc., rekrutiert derzeit 160 Personen für die Studie. In einer weiteren klinischen Studie der Phase III wird der MDM2-Inhibitor Milademetan im Vergleich zu Trabectedin, einem Blocker des onkogenen Transkriptionsfaktors FUS-CHOP, bei MDM2-überexprimierenden ALT/WDL und DDL untersucht. Milademetan hat eine überschaubare Toxizität und eine gewisse Aktivität gezeigt, die zu einer stabilen Erkrankung und/oder einigen partiellen Reaktionen bei DDL führte. ⓘ
Myxoides Liposarkom
Präsentation
Das myxoide Liposarkom (MLS), zu dem auch das so genannte Rundzell-Liposarkom gehört, macht etwa 30 % aller Liposarkome aus. Die Inzidenz erreicht ihren Höhepunkt im vierten und fünften Lebensjahrzehnt, wobei in den meisten Studien die Männer überwiegen. Obwohl es bei Kindern und Jugendlichen selten vorkommt, ist das MLS die häufigste Liposarkomform, die in diesen Altersgruppen diagnostiziert wird. Das MLS präsentiert sich typischerweise als große (1 bis 39 cm; durchschnittlich 12 cm), bewegliche, gut umschriebene, schmerzlose Masse, die sich zwischen einer Woche und 15 Jahren vor der Diagnose entwickelt hat. MLS-Tumore befinden sich in tief liegenden Weichteilen der Oberschenkel (65-80 % der Fälle), Unterschenkel (10-15 % der Fälle), Retroperitoneum (8 % der Fälle) und Arme (5 % der Fälle). In etwa einem Drittel der Fälle metastasieren diese Tumoren in andere Weichteilgewebe (z. B. Retroperitoneum, Thorax oder andere Extremitäten), in das Knochenskelett und/oder die Lunge. Es wird empfohlen, die Patienten bei der Vorstellung auf Knochenmetastasen mittels medizinischer Bildgebung, einschließlich Röntgenaufnahmen, CT-Scans und/oder Magnetresonanztomographie, zu untersuchen. ⓘ
Pathologie
Histopathologische Analysen von MLS (siehe Abb. 3 und 4 im Abschnitt Histopathologie von Liposarkomen) zeigen Zellen, die in einer myxoiden Matrix verstreut sind (d. h. ein Bindegewebshintergrund, der blauer oder violetter erscheint als die rote Farbe von normalem Bindegewebe, wenn diese Gewebe ordnungsgemäß präpariert, H&E-gefärbt und mikroskopisch betrachtet werden). Bei diesen Zellen handelt es sich um Lipoblasten, von denen einige siegelringförmig (eine Form, die darauf hindeutet, dass die Zelle neoplastisch sein könnte), oval oder rund geformt sind. MLS-Tumoren können hyperzellulär sein und solide Platten mit runden Zellen enthalten, die mindestens 5 % aller Zellen ausmachen, oder sie können eine geringe Zellularität aufweisen, die mit Zellen mit blassen Kernen und < 5 % runden Zellen vor einem Hintergrund gekrümmter Kapillaren, die einem Hühnerdrahtmuster ähneln, bevölkert ist. Tumore, die mindestens 5 % runde Zellen enthalten, werden als hochgradig eingestuft, während Tumore mit < 5 % runden Zellen als niedriggradig eingestuft werden. Hochgradige MLS-Tumoren nehmen in der Regel einen aggressiveren klinischen Verlauf als niedriggradige MLS-Tumoren. ⓘ
Genetik
MLS-Tumorzellen werden praktisch durch die Expression eines FUS-DDIT3-Fusionsgens (auch als chimäres Gen bezeichnet) definiert, das in >95 % der Fälle vorkommt, oder eines EWSR1-DDIT3-Fusionsgens, das in den verbleibenden <5 % der Fälle auftritt. Das FUS-DDIT3-Fusionsgen entsteht durch eine Translokation (t(12:16)(q13:p11)) zwischen dem Ort des DDIT3-Gens an Band 12 des q-Arms von Chromosom 12 und dem Ort des FUS-Gens an Band 11 des kurzen Arms von Chromosom 16 (auch p-Arm genannt). Das Fusionsprotein (auch chimäres Protein genannt) dieses chimären Onkogen-Gens, FUS-DDIT3, ist dafür bekannt, dass es die Fettzellreifung hemmt und Neoplasien fördert. Das EWSR1-DDIT3-Fusionsgen (als t(12;22)(q13;q12) bezeichnet) resultiert aus einer Translokation des EWSR1-Gens, das sich in Band 12.2 auf dem q-Arm von Chromosom 22 befindet, mit dem DDIT2-Gen. Das Fusionsproteinprodukt des EWSR1-DDIT3-Gens fördert ebenso wie das FUS-DDIT3-Fusionsprotein die Neoplasie. Trotz dieser Fusionsgenbeziehungen sind weitere Studien erforderlich, um ihren Beitrag zur Entwicklung und/oder Aufrechterhaltung von MLS-Tumoren zu definieren. ⓘ
Diagnose
Niedriggradige und mittelgradige MLS-Tumoren lassen sich histologisch durch ihre klassische Morphologie mit ausgeprägtem Hühnerdraht-Gefäßsystem erkennen, das in einem myxoiden Stroma verstreut ist. Hochgradige MLS-Tumoren können jedoch schwer von anderen Rundzellneoplasien zu unterscheiden sein, insbesondere hochgradige MLS-Tumoren, die eine diffuse Zellmorphologie und/oder eine reine Rundzellmorphologie in einem solchen Ausmaß aufweisen, dass dieses klassische vaskulär-myxoide Muster verdeckt wird. Der Nachweis eines DDIT3-Gen-Rearrangements mit dem FUS- oder EWSR1-Gen durch In-situ-Hybridisierung oder Immunhistochemie oder der RNA-Fusionstranskripte dieser Gene durch Echtzeit-Polymerase-Kettenreaktionen bestätigt die Diagnose hochgradiger sowie unklarer Fälle niedriggradiger oder mittelgradiger MLS-Tumoren. ⓘ
Behandlung und Prognose
MLS wird in der Regel durch chirurgische Resektion behandelt, kann aber auch radikalere Eingriffe erfordern, z. B. die Amputation von Gliedmaßen, wenn das neurovaskuläre Bündel einer Gliedmaße beeinträchtigt ist. Das postoperative Rezidivrisiko innerhalb von 3 Jahren nach der Operation wird mit ~15 % angegeben, wenn nicht der gesamte Tumor entfernt wurde, und ~10 %, wenn der Tumor vollständig entfernt wurde. Die Ergänzung der chirurgischen Resektion durch eine Strahlentherapie hat die lokale Kontrolle von MLS-Tumoren verbessert und wird für die Behandlung inoperabler und rezidivierender MLS empfohlen. Es sind jedoch weitere Studien erforderlich, um den Wert der Strahlentherapie bei der Behandlung der verschiedenen MLS-Varianten zu ermitteln. Chemotherapieschemata mit Ifosfamid, einem Anthrazyklin wie Daunorubicin, Dacarbazin und/oder Trabectedin haben sich als nützlich erwiesen: In einer klinischen Studie der Phase III betrug die progressionsfreie Überlebenszeit bei MLS-Patienten, die mit Trabectedin oder Dacarbazin behandelt wurden, 5,6 bzw. 1,5 Monate. Im Jahr 2015 hat die Food and Drug Administration Trabectedin für die Behandlung von inoperablen und metastasierten Liposarkomen zugelassen. ⓘ
Insgesamt liegt die 10-Jahres-Überlebensrate von MLS-Patienten bei 77 %, eine Überlebensrate, die deutlich höher ist als bei anderen Liposarkomformen. Im Vergleich zu MLS mit niedrigem Risiko sind MLS mit hohem Risiko (definiert durch den Gehalt an Rundzellen im Tumor und/oder andere ungünstige prognostische Indikatoren) mit einer höheren Metastasierungsrate und daher einer kürzeren Überlebenszeit verbunden. Eine zunehmende Tumorgröße (≥ 10 cm) ist stark mit einem MLS höheren Grades und damit einer kürzeren Überlebenszeit verbunden. Weitere Faktoren, die mit ungünstigen Ergebnissen bei MLS in Verbindung gebracht wurden, sind das Vorhandensein von Tumornekrosen, ein Alter von mehr als 45 Jahren, eine Überexpression des P53-Gens und das männliche Geschlecht. Die rundzellige Form der myxoiden Liposarkome scheint ebenfalls eine relativ schlechte Prognose zu haben: In verschiedenen retrospektiven Übersichten wurde festgestellt, dass myxoide Liposarkome in der Regel niedriggradig sind und daher relativ gut auf eine Chemotherapie ansprechen, während hochgradige (d. h. rundzellige) myxoide Liposarkome eine höhere Metastasierungsrate aufweisen, sich aggressiver verhalten und nicht gut auf eine Chemotherapie ansprechen. Es ist jedoch wichtig zu erwähnen, dass fast alle Fälle von myxoiden Liposarkomen bei pädiatrischen Patienten eine ausgezeichnete Prognose hatten. ⓘ
Neuartige Therapien
Ein PPAR-γ-Agonist (d. h. Aktivator), Efatutazon, wurde in einer kleinen Phase-I-Studie an Patienten mit verschiedenen bösartigen Tumoren im fortgeschrittenen Stadium untersucht. Das Medikament führte bei einer Person mit MLS zu einer deutlich dauerhaften Reaktion, was darauf schließen lässt, dass PPAR-γ-Agonisten für die Behandlung dieser Krankheit nützlich sein könnten. Eine in Italien durchgeführte klinische Studie der Stufe II untersucht die Auswirkungen von Trabectedin plus Pioglitazon (einem weiteren PPAR-γ-Agonisten) bei Personen mit stabilen MLS-Tumoren. Die Studie umfasst zwei aufeinander folgende Schritte. Im ersten Schritt wird das Ansprechen der Patienten untersucht, die mindestens 4 Zyklen lang mit Trabectedin allein behandelt wurden. Wenn ein stabiler Krankheitsverlauf erreicht wird, werden in der zweiten Phase die Auswirkungen einer weiteren Behandlung von Patienten, die zunächst auf die Behandlung ansprachen, mit einer Kombination aus Trabectedin und Pioglitazon untersucht. Eine klinische Studie der Phase II steht kurz vor dem Abschluss, um die Wirksamkeit von Sirolimus (einem MTOR-Inhibitor; Sirolimus ist auch als Rapamycin bekannt) plus Cyclophosphamid (einem Chemotherapeutikum) bei metastasiertem oder inoperablem MLS zu untersuchen. In einer klinischen Studie der Phase II werden Patienten rekrutiert, um Sintilimab (ein menschlicher monoklonaler IgG4-Antikörper, der gegen das auf der Zelloberfläche befindliche programmierte Zelltodprotein 1 gerichtet ist) in Kombination mit zwei Chemotherapeutika, Doxorubicin und Ifosfamid, als Erstlinienbehandlung von Weichteilsarkomen einschließlich MLS zu untersuchen. ⓘ
T-Zellen wurden gentechnisch so verändert, dass sie sich gegen das MAGE-A4-Antigen richten, das auf einem HLA-A*02 MAGE-A4-haltigen Peptid exprimiert wird, das sich auf der Oberfläche der neoplastischen Zellen in bestimmten Tumorarten befindet. Diese gentechnisch veränderten Zellen (ADP-A2M4-T-Zellen genannt) griffen verschiedene kultivierte menschliche Krebszellen an, die dieses Antigen trugen, und töteten sie ab. In einer klinischen Studie der Phase 1 ließen sie verschiedene solide Tumorarten bei Patienten schrumpfen, deren Tumoren neoplastische Zellen enthielten, die dieses Antigen exprimierten. Für eine klinische Studie der Phase II werden derzeit Probanden rekrutiert, um die Wirksamkeit und Sicherheit von ADP-A2M4-T-Zellen (die aus den eigenen T-Zellen des Empfängers hergestellt werden) bei HLA-A*02-positiven Patienten mit metastasierten oder inoperablen, MSGE-4-positiven MLS-Tumoren im fortgeschrittenen Stadium zu untersuchen. ⓘ
Pleomorphes Liposarkom
Präsentation
Pleomorphe Liposarkome (PLS), die 5 bis 10 % aller Liposarkomfälle ausmachen, sind schnell wachsende, meist große (>5 cm) und schmerzlose, aber hochgradig bösartige Adipozytentumore. Sie treten hauptsächlich bei Personen im Alter von über 50 Jahren auf, wobei Frauen überwiegen. PLS-Tumore werden selten bei Kindern gefunden. PLS-Tumoren treten in einem Bein oder Arm (65 % der Fälle), im Retroperitoneum oder im Bauchraum (15 % der Fälle) oder in seltenen Fällen in der Rumpfwand, im Samenstrang, im Kopf- und Halsbereich, in der Brustwand, im Beckenraum, im Lungenfell, im Herzbeutel und in der Wirbelsäule auf. Diese Tumoren sind in der Regel in den tiefen Weichteilen lokalisiert, nur 25 % der Fälle treten im subkutanen Gewebe auf. Seltene Fälle von PLS traten bei Personen mit dem Li-Fraumeni- oder Muir-Torre-Syndrom auf, zwei genetisch bedingten Erbkrankheiten, die die Betroffenen für die Entwicklung verschiedener Krebsarten prädisponieren. ⓘ
Pathologie
Die Histopathologie von PLS-Tumoren besteht häufig aus Bereichen, die einem myxoiden Liposarkom ähneln, gemischt mit Bereichen, die undifferenzierte Zellen enthalten. Diese Tumore sind deutlich hyperzellulär und enthalten zumindest einige unterschiedlich geformte Lipoblasten, die pleomorphe Kerne haben. Bereiche mit Nekrose sind häufig, Riesenzellen, von denen einige vielkernig sind und/oder verschlungene Neutrophile enthalten, sind gelegentlich vorhanden, und hyaline Tröpfchen können in einigen Zellen sowie extrazellulär im gesamten Tumor verstreut gesehen werden. Die undifferenzierte Komponente dieser Tumoren besteht meist aus spindelförmigen Zellen, wobei in 25 % der Fälle Zellen mit einer epitheloiden Zellmorphologie zu finden sind. Diese Tumoren weisen zumindest einige Herde auf, deren Histopathologie den hochgradigen Myxofibrosarkom-Histiozytomen ähnelt, einem Tumor, der früher als malignes myxoides fibröses Histiozytom bezeichnet wurde. ⓘ
Genetik
Die neoplastischen PLS-Zellen weisen verschiedene Gen- und Chromosomenanomalien auf: Das TP53-Gen ist in 17-60 % der Fälle deletiert oder mutiert, das RB1-Gen ist in 60 % der Fälle deletiert, und das Neurofibromin-1-Gen geht in 8 % der Fälle durch inaktivierende Mutationen oder in selteneren Fällen durch eine Deletion in der Nähe seiner Position in Band 11.2 auf dem langen Arm von Chromosom 12 verloren. Diese Zellen können auch Zuwächse im genetischen Material um die Bänder 12-15 auf dem kurzen Arm von Chromosom 5, Band 21 auf dem kurzen Arm von Chromosom 1 und Band 22 auf dem langen Arm von Chromosom 7 aufweisen. Die durch diese Anomalien hervorgerufenen Veränderungen der Genkopienzahl ähneln denen, die beim Myxofibrosarkom-Typ der Histiozytome auftreten. Die Rolle(n) dieser Veränderungen in der Genkopienzahl bei der Förderung des PLS ist nicht geklärt. Somit unterscheidet sich das PLS von anderen Liposarkomen dadurch, dass seine neoplastischen Zellen ein komplexes Genom ohne charakteristische genomische Veränderungen oder identifizierbare Gene, die die Krankheit fördern, aufweisen. Der Nachweis von Veränderungen in der Expression der Gene TP53, RB1 und Neurofibromin 1 sowie anderer, bei PLS weniger häufig veränderter Gene (z. B. PIK3CA, Tyrosin-Protein-Kinase SYK, PTK2B, EPHA5 und ERBB4) kann dazu beitragen, einen Tumor als PLS zu definieren, ist aber nicht eindeutig. Eine Verlängerung der Chromosomen-Telomere durch pathologische Mechanismen, die als alternative Telomerverlängerung bezeichnet wird, tritt in den neoplastischen Zellen von ~80 % der PLS-Fälle auf, ist aber bei den anderen vier Formen von Liposarkomen weitaus seltener oder gar nicht zu beobachten. ⓘ
Diagnose
Die Diagnose des PLS hängt von seinem Erscheinungsbild, der Histopathologie und der Genetik ab. Die Histopathologie des PLS ähnelt häufig der des Myxofibrosarkoms, unterscheidet sich aber von diesem Tumor durch seinen Gehalt an pleomorphen Lipoblasten. ⓘ
Behandlung und Prognose
Die radikale chirurgische Resektion ist die Hauptbehandlung des PLS; sie ist auch ein wichtiger palliativer Eingriff zur Linderung der Symptome, die durch die Kompression von Organen und Geweben entstehen. Der chirurgische Eingriff kann die Entfernung eines gesamten komprimierten Organs wie der Niere oder des Dickdarms erfordern. Unabhängig von dieser Operation ist die lokale Rezidivrate jedoch sehr hoch. Der Einsatz von Chemo- und/oder Strahlentherapie in Verbindung mit einer radikalen Operation verlängert das Überleben nachweislich nicht und gilt als umstrittene Maßnahme. Das National Comprehensive Cancer Network empfiehlt zur Behandlung von Personen mit lokalisiertem PLS mit hohem Risiko eine vollständige chirurgische Resektion, wenn möglich in Kombination mit einer Strahlentherapie. Personen mit metastasierter Erkrankung werden mit einer Chemotherapie (z. B. Doxorubicin plus Ifosfamid oder Eribulin) behandelt, die den bei dedifferenzierten Liposarkomen verwendeten Therapien ähnelt (siehe obigen Abschnitt über die Behandlung dieses Liposarkomtyps). Etwa 20 % der PLS-Tumoren metastasieren in entfernte Bereiche, wobei die häufigsten davon die Lunge (82 % der Metastasen), die Leber (18 % der Metastasen) und die Knochen oder die Bauchspeicheldrüse (18 % der Metastasen) sind. Die PLS-Überlebensraten nach 1, 3 und 5 Jahren liegen bei 93 %, 75 % bzw. 29 %. Tumore, die in der Mitte des Rumpfes liegen, größer als 10 cm sind, tief sitzen oder Nekrosebereiche aufweisen, haben eine schlechtere Prognose. ⓘ
Myxoides pleomorphes Liposarkom
Das myxoide pleomorphe Liposarkom (ursprünglich als pleomorphes myxoides Liposarkom bezeichnet) wurde erstmals 2009 in einer großen Studie über Liposarkome beschrieben. Während es zunächst als eine mögliche Variante der myxoiden Liposarkome mit pleomorphen Merkmalen angesehen wurde, stufte die Weltgesundheitsorganisation (2020) es als eine neue und eigenständige Form der Liposarkome ein. Diese Einstufung beruhte auf der Feststellung, dass die myxoiden pleomorphen Liposarkome zwar histopathologische Merkmale aufweisen, die denen der myxoiden Liposarkome ähneln, dass sie sich aber klinisch und vor allem durch kritische genetische und molekulare Merkmale von den myxoiden und den anderen drei Liposarkomformen unterscheiden. ⓘ
Präsentation
Das myxoide pleomorphe Liposarkom (MPL) ist eine außerordentlich seltene und hochaggressive Form der Liposarkome, die bei Kindern, Jugendlichen, jungen Erwachsenen und - in einer neueren Studie - bei Personen über 50 Jahren auftritt. MPL-Tumore treten als tiefe Weichteilmassen auf, die häufig im Mittelfell und seltener in den Extremitäten, im Kopf- und Halsbereich, in der Bauchhöhle oder im Rumpf lokalisiert sind. Mindestens zwei Fälle von MPL traten bei Personen mit dem Li-Fraumeni-Syndrom auf, einer vererbten genetischen Störung, die eine Prädisposition für die Entwicklung verschiedener Krebsarten darstellt. ⓘ
Pathologie
Bei der histopathologischen Analyse bestehen die MPL-Tumoren aus Bereichen, die einem herkömmlichen myxoiden Liposarkom ähneln; diese Bereiche, die 30-50 % der gesamten Tumorfläche ausmachen, weisen eine reichhaltige myxoide Matrix, eine gut entwickelte Kapillarvaskulatur, blande Zellen, die rund und/oder leicht spindelförmig sind, vakuolisierte Lipoblasten und vielkernige Zellen in Form kleiner Blumen auf. In diesen Bereichen finden sich jedoch auch vereinzelt stark pleomorphe Zellen, die einen größeren Grad an Kernvergrößerung und Unregelmäßigkeit aufweisen als die Zellen in myxoiden Liposarkomtumoren. Andere Bereiche von MPL-Tumoren sind eher zellulär und bestehen aus schnell wachsenden und stark pleomorphen Lipoblasten. ⓘ
Genetik
Die neoplastischen Zellen in MPL exprimieren nicht die FUS-DDIT3- oder EWSR1-DDIT3-Fusionsgene, die von den neoplastischen Zellen in >95% bzw. <5% der Fälle von myxoiden Fibrosarkomen exprimiert werden. Eine Inaktivierung des Tumorsuppressorgens RB1 durch Deletion oder pathologische Suppression findet sich in allen MPL-Fällen. Neoplastische MPL-Zellen weisen häufig auch andere Veränderungen in ihren Chromosomen auf. Sie können einen anormalen Zuwachs an genetischem Material aufweisen, das normalerweise auf den Chromosomen 1, 6, 7, 8, 19, 21 und/oder X zu finden ist, und Verluste an genetischem Material, das normalerweise auf den Chromosomen 2, 3, 4, 5, 10, 11, 12, 13, 14, 15, 16, 17 und/oder 22 zu finden ist. Das in Band 14 auf dem langen Arm von Chromosom 13 verlorene genetische Material umfasst nicht nur das RP1-Gen, sondern auch die Gene RCBTB2, DLEU1 und ITM2B. Aufgrund ihrer Seltenheit und der erst kürzlich erfolgten Definition sind die molekularen Merkmale und die Bedeutung dieser genetischen Anomalien noch nicht vollständig geklärt. Dennoch lassen Studien vermuten, dass Verluste in einem oder mehreren der RB1-, RCBTB2-, DLEU1- und ITM2B-Gene, insbesondere aber im RP1-Gen, zur Entstehung und/oder zum Fortschreiten der MPL beitragen können. ⓘ
Diagnose
Die Diagnose von MPL hängt vom klinischen Erscheinungsbild der Tumoren, der histopathologischen Ähnlichkeit mit myxoiden Liposarkomen und vor allem vom Fehlen der FUS-DDIT3 sn EWSR1-DDIT3 Fusionsgene in den neoplastischen Zellen ab. ⓘ
Behandlung und Prognose
Während bei Patienten mit MPL eine chirurgische Resektion zur Entfernung des Tumors durchgeführt wurde, ergab eine Untersuchung aus dem Jahr 2021, dass es keine einheitlichen Empfehlungen für die Standardbehandlung von MPL im Hinblick auf Bestrahlung und Chemotherapie (entweder allein oder in Kombination mit einer Operation) zur Behandlung dieser Tumoren gibt. ⓘ
Histopathologie von Liposarkomen
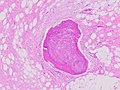
Abb. 1 Mikroskopische Aufnahme der Knochenbildung in einem Liposarkomtumor

Abb. 2 Mikroskopische Aufnahme eines entdifferenzierten Liposarkomtumors

Abb. 3 Mikroskopische Aufnahme eines myxoiden Liposarkomtumors in niedrigerer Auflösung

Abb. 4 Mikroskopische Aufnahme eines myxoiden Liposarkomtumors mit höherer Auflösung ⓘ


.jpg)
.JPG)
Medizinische Bildgebung
Medizinischer Ultraschall und Magnetresonanztomographie (MRT) sind bei Liposarkomen hilfreich und oft unerlässlich, um ihre Ausdehnung, die chirurgische Zugänglichkeit und die Beziehung zu beobachteten Organstörungen zu bestimmen. Da die Ultraschalluntersuchung in der Regel nicht in der Lage ist, ein Liposarkom von einem gutartigen Lipom zu unterscheiden, ist die MRT die erste bildgebende Methode der Wahl, um diese Unterscheidung zu belegen. ⓘ
Bei einem myxoiden Liposarkom zeigt sich auf T1-gewichteten MRT-Bildern eine Masse mit geringer Signalintensität und Herden mit hoher Signalintensität. Die Masse zeigt eine hohe Signalintensität auf T2-gewichteten Bildern. Dies ist darauf zurückzuführen, dass sie überwiegend mukoide Substanz (niedrige Signalintensität bei T1) und eine geringe Menge reifen Fetts (hohe Signalintensität bei T1) enthält. Die Masse ist gut abgegrenzt, lobulär, multilokulär oder oval geformt, ohne Infiltration in die umliegenden Strukturen. ⓘ

Abb. 5 Ultraschallbild eines Liposarkoms mit echoreichen Bereichen, die von der lipomatösen Matrix reflektiert werden, und echoarmen Bereichen, die von den nicht lipomatösen Bereichen reflektiert werden.
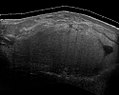
Abb. 6 Ultraschalluntersuchung eines Liposarkoms, das ein Lipom imitiert. Diese homogene Masse mit hohem Echo hat das gleiche Aussehen wie ein Lipom.

Abb. 7 MRT eines myxoiden Liposarkoms hohen Grades in der linken Axillarregion eines 40-jährigen Mannes, hervorgehoben durch seine weiße Farbe in diesem horizontalen Schnitt durch den Tumor. ⓘ



Gesellschaft und Kultur
Bemerkenswerte Fälle
- Chad Brown (1961-2014), ein Pokerspieler, starb an einem Liposarkom
- Richard Feynman (1918-1988), ein theoretischer Physiker, starb nach einer Operation zur Behandlung der Krankheit.
- Rob Ford (1969-2016), ehemaliger Bürgermeister von Toronto und Stadtrat von Toronto, starb an einem pleomorphen Liposarkom.
- Hokie Gajan (1959-2016), ehemaliger Running Back der New Orleans Saints und Radiokommentator für das Team, starb an einem Liposarkom.
- Charlie Davies (1986-), ehemaliger Fußballspieler für die Philadelphia Union in der Major League Soccer, bei dem 2016 ein Liposarkom diagnostiziert wurde.
- Mark Strand (1934-2014), ehemaliger US-Poet Laureate und Pulitzer-Preisträger, starb an einem Liposarkom. ⓘ
Epidemiologie
Die Inzidenz des Liposarkoms wird international mit etwa 2,5 Neuerkrankungen je einer Million Einwohner und Jahr angegeben. Mit einem mittleren Erkrankungsalter von 50 Jahren handelt es sich um einen Tumor des Erwachsenen, der gleichwohl selten auch bei Kindern und jungen Erwachsenen beobachtet wird. Männer sind geringfügig häufiger betroffen als Frauen. Geographische oder ethnische Häufigkeitsunterschiede wurden bislang nicht berichtet. ⓘ
Ätiologie
Die der Entstehung eines Liposarkoms zugrunde liegenden Ursachen sind weitgehend ungeklärt. Beschrieben wird eine mögliche Beziehung zu vorausgegangenen Verletzungen und einer Exposition gegenüber ionisierender Strahlung. Das Lipom, ein gutartiger und ungleich häufigerer Fettgewebstumor, ist keine typische Vorläuferveränderung des Liposarkoms, soll aber laut einigen Autoren in Einzelfällen dessen Ausgangspunkt bilden können. Andere Quellen bestreiten diese Ansicht und verweisen darauf, dass ein Übergang eines Lipoms in ein Liposarkom bislang nie überzeugend dokumentiert werden konnte. ⓘ
Klinische Symptomatik
Liposarkome werden häufig erst in fortgeschritteneren Stadien als tief gelegene, langsam wachsende tumoröse Gewebsmasse klinisch auffällig. Die genaue Symptomatik wird dabei vorwiegend von der Lokalisation des Tumors bestimmt. Mit dem Tumorwachstum möglicherweise einhergehende Allgemeinerscheinungen sind zum Beispiel Müdigkeit, Abgeschlagenheit, Gewichtsverlust, Übelkeit und Erbrechen. ⓘ
Diagnose
Bildgebende Verfahren wie die Computertomographie, die Magnetresonanztomographie, die Angiographie oder die Szintigraphie liefern diagnostische Hinweise und ermöglichen eine Einschätzung der Ausbreitung des Tumorleidens. Zur definitiven Diagnosesicherung ist in der Regel eine Biopsie und die histologische Untersuchung des gewonnenen Tumorgewebes durch einen Pathologen erforderlich. ⓘ
Therapie
Der erfolgversprechendste therapeutische Ansatz ist die vollständige chirurgische Entfernung des Tumors unter Einhaltung eines ausreichenden Sicherheitsabstandes. Weitere Therapieoptionen sind die lokale Bestrahlung und eine Chemotherapie. Obwohl das Liposarkom als das strahlensensibelste Sarkom gilt, konnte eine Steigerung der Überlebenszeit durch eine Radiotherapie in wissenschaftlichen Studien bislang nicht überzeugend gezeigt werden. Auch die Chemotherapie des Liposarkoms hat gegenwärtig noch experimentellen Charakter. ⓘ
Prognose
Die Heilungsaussichten sind neben der Möglichkeit einer kompletten chirurgischen Entfernung davon abhängig, welcher feingewebliche Subtyp des Liposarkoms vorliegt. Die gut differenzierten sowie die meisten myxoiden Liposarkome zeigen mit einer Fünf-Jahres-Überlebensrate von 100 beziehungsweise 88 Prozent eine günstige Prognose. Diese ergibt sich unter anderem daraus, dass diese Formen kaum zu Metastasenbildung neigen. Hingegen versterben etwa 50 Prozent der Patienten mit einem rundzelligen oder schlecht differenzierten Liposarkom binnen fünf Jahren an ihrem Tumorleiden. Metastatische Tumorabsiedlungen betreffen vor allem die Lunge (20 %), Knochen (8 %), Lymphknoten (6 %) und die Leber (5 %). ⓘ